
Translate this page into:
Breaking new ground: Exploring de novo chromosomal rearrangements in 1p36 microdeletion
Address for correspondence: Raniah S. Alotibi, Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, King Saud bin Abdulaziz University for Health Sciences, P.O. Box 3660 Riyadh 11481, Riyadh, Saudi Arabia. Phone: +966-1144299999. E-mail: raniao@ksau-hs.edu.sa
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Qassim Uninversity and was migrated to Scientific Scholar after the change of Publisher.
Abstract
ABSTRACT
Chromosomal structural variations (SVs) are linked to a wide range of phenotypes and arise due to disruptions during DNA replication, which can affect gene function within the SV regions. This case report details a patient diagnosed with neurodevelopmental delay. Detailed investigation through array comparative genomic hybridization revealed two pathogenic SVs on chromosome 1, which align with a 1p36 microdeletion, and a microduplication at 2p35.3, the latter being classified as a variant of unknown significance. The patient’s clinical presentation is consistent with the 1p36 deletion syndrome, characterized by specific developmental delays and physical anomalies. Further genetic analysis suggests that these terminal rearrangements might stem from an unbalanced translocation between the short arms of chromosomes 1 and 2. This case underscores the complexity of interpreting multiple concurrent SVs and their cumulative effect on phenotype. Ongoing research into such chromosomal abnormalities will enhance our understanding of their clinical manifestations and guide more targeted therapeutic strategies.
Keywords
1p36 deletion syndrome
2p25.3 duplication
case report
chromosomal rearrangement
chromosome
unbalanced translocation

Introduction
Chromosomal structural variation (SV) is associated with various phenotypes.[1-3] Usually, SV occurs during DNA replication processes, repair, or recombination.[4] This rearrangement reflects on the phenotype due to gene rupture change in the dosage of genes, or it effects gene fusion.[1,4,5]
1p36 microdeletion syndrome (MIM 607872) is a common deletion syndrome in the United States of America, occurring in one in every 5000 individuals.[6] Terminal rearrangements of chromosome 1 account for 67% of all the SVs within this chromosome and 20% of all unbalanced S.Vs. associated with congenital disabilities and intellectual disabilities.[7,8] This region has variable sizes of breakpoints, ranging from 1 to 12.9 Mb.[9,10] The phenotype of 1p36 deletion syndrome varies depending on the deletion size. Moreover, it may be associated with clinical features, including global developmental delay, intellectual disability, congenital hypotonia, facial dysmorphism, epilepsy, hearing defects, cardiac malformation, microcephaly, and growth defects.[11,12] The discrepancies between patients are associated with genotype and phenotype correlation, which is linked to the size and genes affected.[13,14]
No literature has explained the microduplication in 2p25.3 in detail except for one study that correlated the presence of microduplication within 2p25.3 with childhood-onset schizophrenia.[15]
Herein, we have described a case report associated with a 1p36 deletion, a well-known syndrome, and a 2p25.3 microduplication, an unknown one. Finding a 1p36.33p36.32 deletion and a 2p25.3 microduplication in one patient may be associated with further complications or overlapping phenotypes. We explored the clinical manifestations associated with these genotypes. We delineated the 1p36 deletion phenotype and compared it with that of the previously published probands.
Methods
This retrospective study was performed by recruiting the patient’s basic information, phenotype, family history, consanguinity, clinical investigations, and genetic testing. A 4-year-old girl who has global developmental delay and dysmorphic features was seen at the National Guard Hospital (NGH), Riyadh, Kingdom of Saudi Arabia. Blood samples were taken from the proband and her parents.
Ethical approval
The ethical approval was obtained from King Abdullah International Medical Research Center (KAIMRC) in Riyadh, Saudi Arabia. The ethics committee’s approval was received with I.D. number S.P. 19/161/R.
Case Presentation
A 4-year-old girl was born to 3-degree sister parents with three healthy offspring [Figure 1]. The pregnancy and delivery histories were uneventful. The proband was born at term, with a median birth weight of 3 kg (median). At the age of 2 months, she experienced one episode of a suspected seizure that resolved spontaneously. The suspected seizure was in the form of facial twitching, eye-staring, and tonic movements of the four limbs and was associated with crying, followed by a decreased level of consciousness. She had a moderate-to-severe global developmental delay. She sat down at 12 months, stood up at 2 years, and started walking at 2 years. After 4 years, language development was limited to ten isolated words. She was unable to write sentences. At 3 years old, she had difficulty understanding simple commands. She was unable to use the toilet, and she was using a diaper. She had hyperactivity, poor eye contact, stereotypies, and repetitive interests such as rocking, switching lights on/off, and playing with a cell phone.
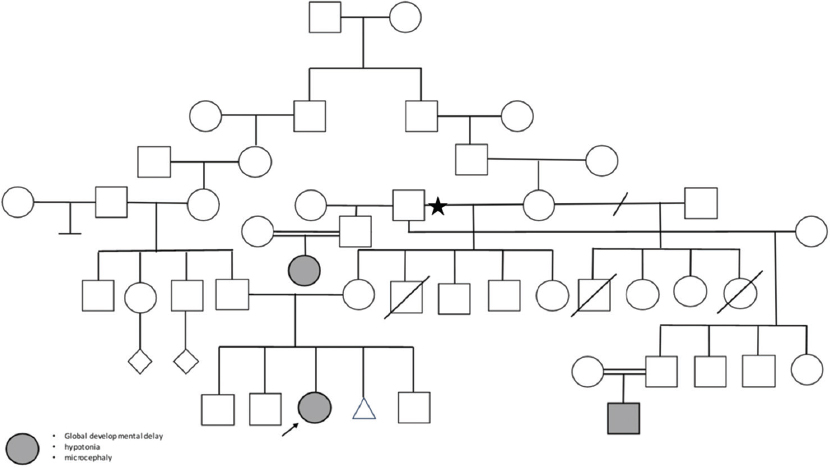
- The pedigree of the consanguine family arrow indicates the proband and the highlighted circle/square indicates the same phenotype in other family members. The three cases are descendent of the same grandfather (marked with a star)
Growth parameters at 4 years were as follows: height, 99 cm (7th percentile); weight, 15.5 kg (50th percentile); and head circumference, 47 cm (−2 SD). Examination revealed the presence of dysmorphic features, including brachycephaly, a deep-seated eye, a prominent nasal bridge, and a smooth philtrum. Neurological examinations revealed wide ataxic gates, axial hypotonia, and peripheral hypertonia. The other examination results were unremarkable. Thyroid-stimulating hormone and free T4 levels were within normal ranges. Metabolic investigations including newborn screening tests and plasma tests for amino acids, ammonia, and lactic acid were normal. Echocardiography revealed no abnormalities. A skeletal survey at 3 years revealed the presence of 11 ribs and mild partial uncovering of the lateral aspect of both femoral epiphyses. Brain computed tomography at 9 months demonstrated the presence of a large anterior fontanel, widening of the bifrontal cerebrospinal fluid (CSF) space, and a persistent small cyst of the septum pellucidum. Brain magnetic resonance imaging at 9 months revealed bilateral frontal atrophy, deep white matter atrophy, prominent CSF space in the frontal region, and Sylvain’s fissures.
Genetics analysis
Chromosomal analysis
Karyotyping was performed on lymphocytes using the phytohemagglutinin-stimulated lymphocyte culture method followed by the G-banding technique.
Array based technology
The blood sample was obtained from the patient, and the DNA was extracted using a DNeasy Blood and Tissue kit (Qiagen, Inc., Valencia, CA, USA). The sample was then sent to a commercial laboratory accredited by the collage of American Pathology for using whole genome oligonucleotide array comparative genomic hybridizations (CGHs) and genotype analysis using a custom-designed single nucleotide polymorphisms (SNP) microarray (GenomeDx v5). The array design is based on the human genome assembly (GRCh37/hg19). Moreover, the result was reported according to the International System for Human Cytogenetic Nomenclature (ISCN) guideline. The copy number variants (CNVs) clinical interpretation was performed according to the recommendations of the American College of Medical Genetics and Genomics. The array contains approximately 118,000 probes that provide CNV and 66,000 probes that generate genotype information through analysis of SNPs. The array detects CNV change of more than 200Kb. On average, across the entire unique sequence of the human genome and between 500 bp and 15 kb, more than 200 targeted regions. The array also detects more than 5 mb homozygosity (ROH) regions. ROH is reported if there is at least one region of more than 10 Mb, or two regions, each more than 8 Mb, suggesting identity by descent. The possibility of uniparental disomy is reported when there is a terminal ROH of more than 10 Mb or an interstitial ROH of more than 20 Mb. The following are not routinely reported: being and likely benign variant: carrier status for autosomal recessive disorder: duplication <500 kb and deletions smaller than 250 kb in the region outside of targeted areas in a blood specimen: duplication <1 Mb and deletions smaller than 300 kb in the region outside of the targeted areas in an oral tested sample.[16-20]
Results
Blood karyotyping revealed that the presence of 46, XX. De r(1) and (1) (p36.2) was added to the probe. The array-based method illustrated the presence of two de novo CNVs, as indicated in Table 1. This patient has an apparently de novo terminal loss of at least 2.3 Mb extending from cytogenetic band 1p36.33 to 1p36.32 and an apparently de novo gain of at least 1.3 Mb within cytogenetic band 2p25.3. The terminal CNV loss in 1p36.33p36.32 is associated with the 1p36 microdeletion syndrome. In contrast, terminal duplication in 2p.53 is a variant of uncertain significance (VUS). Both parents underwent array CGH, and the results were normal.

The DESIPHER and ClinVar databases illustrate that the 1p36 terminal deletion is a well-recognized syndrome and is correlated with our proband’s phenotype.[16,17] The duplication with the cytogenetic band 2p25.3 was a CNV with VUS. The duplication interval contains a portion of the FAM110C and SNTG2 genes and the centric SH3YL1, ACP1, FAM150B, TMEM18, and LINCO1115 genes, none of which are associated with the known clinical disorders at present.[18] This region is not known to have a high CNV in the normal population.[19] In the DECIPHER database, this CNV showed no association with the syndrome,[17] and further looking into the ClinVar database, pathogenic and likely pathogenic SVs, no matching sizes were observed; however, different SVs with heterozygous inheritance were associated with different phenotypes.[16] None of the morbid genes were affected by this duplication.
Discussion
The constitutional deletion of chromosome 1p36 results in a syndrome with global developmental delay, severe to profound intellectual disability, severe language delay to absent speech, behavioral disorders, seizures, and multiple congenital anomalies (OMIM 607872),[21,22] similar to our proband. Based on the 13 subjects described by Shapira et al. 1997 and 86 probands, the dysmorphic features of the 1p36 syndrome include brachycephaly, large, late-closing anterior fontanel, straight eyebrows, deep-set eyes, a wide/flat nasal bridge, midface hypoplasia, asymmetric/low set ears, and a pointed chin.[23] A few of these dysmorphic features were observed in the proband. Brain atrophy was observed in the proband, as previously reported [Supplementary Table 1].[12] Head circumference 2DS below the mean and short stature were observed in our proband and previously published cases with a 1p36 deletion.[20] Additional clinical characteristics, including cardiomyopathy and hearing impairment, have been reported to be associated with 1p36 microdeletion; however, none were observed in our proband[12] [Supplementary Table 1].
The deletion sizes and locations within the 1p36 region varied. The phenotype severity may be explicit for deletion sizes >10 Mb.[24]
The mechanism of 1p36 deletions varies, with the majority being a pure terminal deletion followed by an interstitial deletion.[24] Terminal deletion of 1p36 due to a derivative of an unbalanced translocation has been observed (8/50)[24] [Supplementary Table 1]. The terminal microdeletion 1p36 and the terminal microduplication in 2p25.3 in our proband could be a derivative of an unbalanced translocation. One of the parents might be a carrier of a balanced translocation between the short arms of chromosomes one and two. However, fluorescence in situ hybridization (FISH) should be performed on the proband, along with FISH and karyotyping of the parents, to confirm our theory.
The 1p36 deletion affected many genes associated with morbid diseases [Table 2]. Among these genes are potassium voltage-gated channel subfamily, a regulatory beta subunit 2 has been associated with epilepsy,[25] and matrix metallopeptidase 23B.[26] In addition, the SKI proto-oncogene (SKI) gene is implicated in cleft palate formation.[26] Neurodevelopmental and neuropsychiatric features have been linked to mutations in the gamma-aminobutyric acid type A receptor subunit theta gene,[27] further supporting the observed phenotype in the proband. **The risk of having a baby with the same phenotype was the same across the population. However, identifying two other members with the same phenotype might be associated with a mutation in this region, resulting in a high risk of deletion. In contrast, microduplication on chromosome 2 has been reported to be associated with developmental delay, which is related to larger duplications and spans morbid genes.[28]
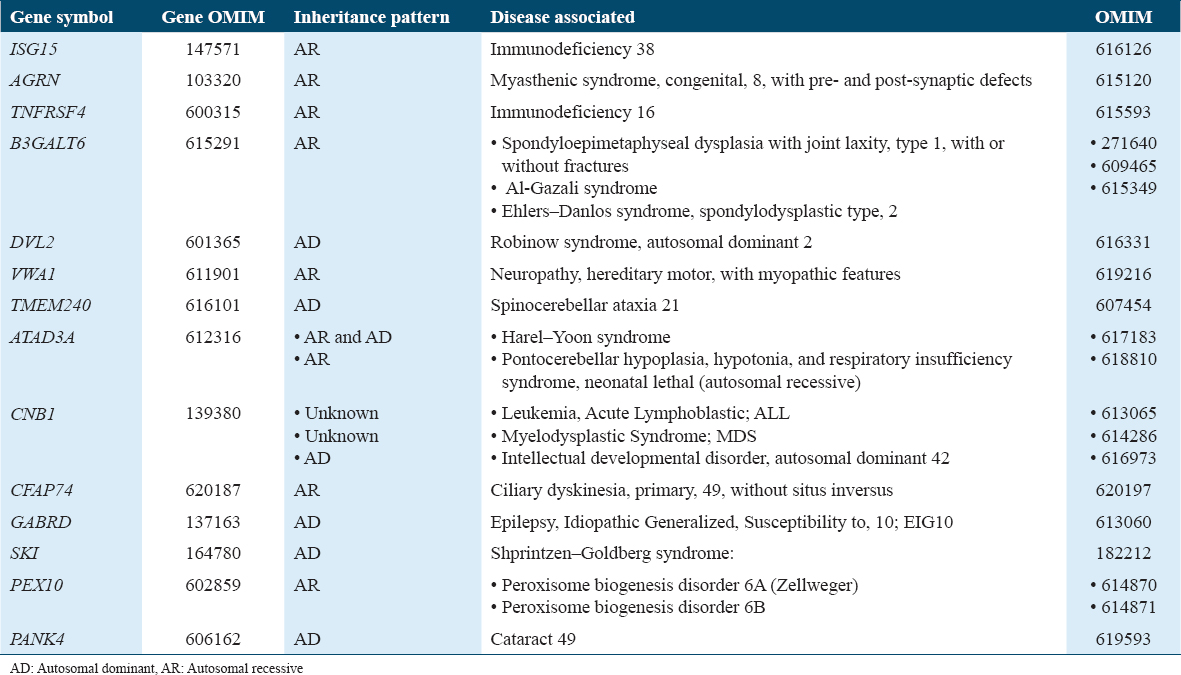
Other studies have correlated the presence of microduplications within 2p25.3 with childhood-onset schizophrenia; however, none of the CNVs mentioned in the study overlapped with our CNV.[15] In our proband, there is no strong evidence that it contributes to the phenotype of microduplication in 2p25.3; therefore, the gain in CNV remains a VUS. We present a case of 1p36 microdeletion of 2.2 Mb that might be an unbalanced translocation derivative. Therefore, we examined the proband by FISH and parents using conventional cytogenetic techniques, including FISH and karyotyping. Subsequently, the possibility of harboring a balanced translocation in the family that yields a derivative of an unbalanced translocation might explain the recurrent phenotype.
The study by Almokali et al., which describes a case of nephrotic syndrome associated with pretibial epidermolysis bullosa due to a CD151 tetraspanin defect, provides valuable insights into the role of membrane proteins in cellular integrity and disease manifestation. CD151, a tetraspanin family protein, is crucial for the stability and function of cell membranes and is involved in cellular processes such as adhesion, motility, and signaling.[29]
In the context of chromosome 1q deletions, where various genes may be deleted or disrupted, understanding the impact of specific protein deficiencies like that of CD151 can provide a model for how similar disruptions might contribute to the broad phenotypic spectrum observed in chromosomal deletion syndromes. For example, if genes coding for proteins involved in similar pathways as CD151 are impacted by the 1q deletion, this could lead to disruptions in cell adhesion and membrane integrity, potentially contributing to some of the developmental and neurological symptoms observed in the syndrome.
Further understanding of the genetic underpinnings of immune system dysfunction in chromosomal deletion syndromes can be gleaned from studies like the one on Apoptotic Protease Activating Factor-1 (APAF-1) and its role in CD4+ depletion during HIV progression. APAF-1, a key component in the apoptotic pathway, has been shown to influence immune cell dynamics significantly, a process that could be disrupted in other genetic conditions involving deletions or duplications.
The observed mechanism in HIV-related immune depletion, where APAF-1 is upregulated leading to increased cell apoptosis, could parallel mechanisms in syndromes like 1p36 deletion where genetic disruptions potentially affect cellular stability and apoptosis pathways. This comparison is particularly pertinent given the neurological and developmental challenges in 1p36 deletion syndrome, where disrupted apoptosis could exacerbate or contribute to the phenotypic severity, similar to immune dysfunction in HIV.
Understanding these parallels can provide a unique insight into how genetic integrity affects cellular functions across different systems and diseases, underscoring the importance of a comprehensive genetic and cellular function assessment in diagnosing and managing complex genetic disorders. Such insights emphasize the need for further research into the specific genes affected by the 1p36 deletion and their roles in cellular function and integrity, potentially revealing targets for therapeutic intervention that could mitigate some of the severe outcomes associated with the syndrome.[30]
In terms of clinical implications, our findings highlight the necessity of comprehensive genetic counseling for families, emphasizing the potential for recurrent phenotypes due to hereditary or de novo genetic alterations. Future research should aim to explore the full spectrum of phenotypic outcomes associated with different sizes and types of deletions within the 1p36 region. In addition, investigating the therapeutic potential of targeting specific pathways disrupted by such deletions could offer new avenues for intervention.
Conclusion
The study of 1p36 deletion syndrome provides crucial insights into the relationship between genomic alterations and phenotypic outcomes. Continued research in this area is vital, with a focus on improving diagnostic techniques and expanding our understanding of genetic disorders, ultimately enhancing patient care and management.
Ethics Approval and Consent to Participate
Approved by the Institutional Review Board at the local Institutional Review Board at King Abdullah International Medical Research Center (KAIMRC) Riyadh, Saudi Arabia. The ethics committee’s approval was received Protocol Approval Number S.P. 19/161/R.
Informed Consent Statement
The patient and family have consented and a written consent form was obtained from them.
Availability of Data and Material
The data presented in the study are deposited in the ClinVar database repository, accession number SCV003803010-SUB12655703.
Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Funding Statement
The authors would like to extend their appreciation to the King Salman Center for Disability Research for funding this work through Research Group no KSRG -2023-543.
Authors’ Contributions
M.A. provides the writing of the paper. R.A. conceptualized the idea, collected data, and reviewed the article. A.A. write the proband case and review the article. M.G.A. reviewed the article. H.A review the article. N.A. and Y.A. collected data and reviewed the article.
Acknowledgments
The authors gratefully acknowledged the patients and the parents involved in this study, including the pediatric clinic at NGH. The authors would like to extend their appreciation to the King Salman Center for Disability Research for funding this work through Research Group no KSRG -2023-543.
References
- Genomic disorders:Structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417-22.
- [Google Scholar]
- Implications of human genome architecture for rearrangement-based disorders:The genomic basis of disease. Hum Mol Genet. 2004;13:R57-64.
- [Google Scholar]
- Human structural variation:Mechanisms of chromosome rearrangements. Trends Genet. 2015;31:587-99.
- [Google Scholar]
- Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet. 2016;17:224-38.
- [Google Scholar]
- Recurrent interstitial 1p36 deletions:Evidence for germline mosaicism and complex rearrangement breakpoints. Am J Med Genet A. 2010;152A:3074-83.
- [Google Scholar]
- Bilateral perisylvian polymicrogyria, periventricular nodular heterotopia, and left ventricular noncompaction in a girl with 10.5-11.1 Mb terminal deletion of 1p36. Am J Med Genet A. 2008;146:2891-7.
- [Google Scholar]
- Further delineation of nonhomologous-based recombination and evidence for subtelomeric segmental duplications in 1p36 rearrangements. Hum Genet. 2009;125:551-63.
- [Google Scholar]
- Physical map of 1p36, placement of breakpoints in monosomy 1p36, and clinical characterization of the syndrome. Am J Hum Genet. 2003;72:1200-12.
- [Google Scholar]
- Electroclinical features of epilepsy associated with 1p36 deletion syndrome:A review. Epilepsy Res. 2018;139:92-101.
- [Google Scholar]
- Identification of proximal 1p36 deletions using array-CGH:A possible new syndrome. Clin Genet. 2007;72:329-38.
- [Google Scholar]
- Further delineation of deletion 1p36 syndrome in 60 patients:A recognizable phenotype and common cause of developmental delay and mental retardation. Pediatrics. 2008;121:404-10.
- [Google Scholar]
- Tiling path resolution mapping of constitutional 1p36 deletions by array-CGH:Contiguous gene deletion or “deletion with positional effect”syndrome? J Med Genet. 2005;42:166-171.
- [Google Scholar]
- Refinement of causative genes in monosomy 1p36 through clinical and molecular cytogenetic characterization of small interstitial deletions. Am J Med Genet A. 2010;152A:1951-9.
- [Google Scholar]
- Microduplications disrupting the MYT1L gene (2p25.3) are associated with schizophrenia. Psychiatr Genet. 2012;22:206-9.
- [Google Scholar]
- 2023. ClinVar Variation Report. Available from: https://www.ncbi.nlm.nih.gov/clinvar/docs/variation_report/#the-clinvar-variation-report
- The diagnostic yield of CGH and WES in neurodevelopmental disorders. Front Pediatr. 2023;11:1133789.
- [Google Scholar]
- 1p36 deletion syndrome:Review and mapping with further characterization of the phenotype, a new cohort of 86 patients. Am J Med Genet A. 2023;191:445-58.
- [Google Scholar]
- Molecular diagnostic yield of whole-exome sequencing in Saudi autistic children with epilepsy. Int J Health Sci (Qassim). 2023;18:15-22.
- [Google Scholar]
- Distal 1p36 deletions:The phenotypic spectrum and molecular characterization. J Med Genet. 1997;34:721-28.
- [Google Scholar]
- Microarray analysis of 50 patients reveals the critical chromosomal regions responsible for 1p36 deletion syndrome-related complications. Brain Dev. 2015;37:515-26.
- [Google Scholar]
- Loss of the potassium channel beta-subunit gene, KCNAB2, is associated with epilepsy in patients with 1p36 deletion syndrome. Epilepsia. 2001;42:1103-11.
- [Google Scholar]
- Delineation of mechanisms and regions of dosage imbalance in complex rearrangements of 1p36 leads to a putative gene for regulation of cranial suture closure. Eur J Hum Genet. 2005;13:139-49.
- [Google Scholar]
- The human gamma-aminobutyric acid A receptor delta (GABRD) gene:Molecular characterization and tissue-specific expression. Gene. 2002;292:25-31.
- [Google Scholar]
- Copy number variations on chromosome 2:Impact on human phenotype, a cross-sectional study. Porto Biomed J. 2023;8:e198.
- [Google Scholar]
- Nephrotic syndrome:Pretibial epidermolysis bullosa in a patient with CD151 tetraspanin defect:A case report. Int J Health Sci (Qassim). 2023;18:35-40.
- [Google Scholar]
- Role of apoptotic protease activating factor-1 in CD4+depletion during HIV progression. Int J Health Sci (Qassim). 2024;18:30-8.
- [Google Scholar]




