
Translate this page into:
Molecular diagnostic yield of whole-exome sequencing in Saudi autistic children with epilepsy
Address for correspondence: Dr. Asmaa Ali Alharbi, Department of Biochemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia. Email: aanalharbi@kau.edu.sa
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Qassim Uninversity and was migrated to Scientific Scholar after the change of Publisher.
Abstract
ABSTRACT
Objectives:
Autism spectrum disorder (ASD) is a neurological condition that affects social communication and causes repetitive behavior. Autistic children often have comorbidities such as epilepsy. Although the co-occurrence of epilepsy and ASD is frequent, the genetic basis for this association is not fully understood. Many cases of ASD and epilepsy remain unresolved without a molecular diagnosis. The purpose of this study was to determine the molecular diagnostic yield in two Saudi families with a single affected offspring with both ASD and epilepsy using whole-exome sequencing (WES).
Methods:
Pediatric patients were diagnosed by a pediatric psychiatrist and neurologist, and diagnosed according to the diagnostic and statistical manual of mental disorders (DSM-V) criteria. WES was used to analyze the coding region of DNA from the two trios. Enrichment analysis was performed on the final list of genes.
Results:
De novo variations were detected in eleven genes (two in ZBTB17 and FRG, and one each in CAD, CTNNA3, GILGA8J, CCZ1, CASKIN1, growth differentiation factor (GDF7), NBPF10, DUX4L4, and ZNF681). Variations in CTNNA3, GOLGA8J, CASKIN1, CCZ1, and NBPF10 genes were correlated to autism. In addition, similar studies found that CAD, CASKIN1, and GOLGA8J were candidate genes for epilepsy. FRG1 and DUX4 variations were associated with facioscapulohumeral muscular dystrophy. The expression of ZBTB17 and GDF was high in nervous system, and variations in these genes might be correlated to autism and epilepsy.
Conclusion:
Not all the genes presumed to cause ASD and epilepsy in this study were previously identified, suggesting that more genes were suspected of being involved in ASD and epilepsy co-occurrence.
Keywords
Autistic spectrum disorder
epilepsy
whole-exome sequencing
De novo variations
Saudi families

Introduction
Autism spectrum disorder (ASD) is one of the most common and complex childhood neurodevelopmental condition that is typically associated with comorbidities of a psychological or physiological nature.[1,2] Most common comorbidities include anxiety, depression, attention-deficit/hyperactivity disorder, epilepsy, gastrointestinal problems, sleep disorders, eating disorders, learning disability, intellectual disability, and obsessive-compulsive disorder.[3] A recent estimate from a network funded by the Center for Disease Control shows that around 1 in 44 children are diagnosed with ASD[4] with a higher prevalence in boys than girls.[5]
Epidemiological studies worldwide showed that the prevalence of ASD is increasing,[6,7] with rates ranging from 0.14 to 2.9% in Arab countries (Saudi Arabia, United Arab Emirates, Oman, Kuwait, Qatar, and Bahrain).[8] Studies conducted in various cities across Saudi Arabia have confirmed a rise in the prevalence of ASD.[9-12]
The etiology of ASD has not been fully attributed to a single factor. Several influences seem to be involved in the occurrence of this condition. These involve the epigenetic interactions between genetic and environmental influences. The previous studies assumed that several environmental influences contribute to autism, such as parental age, prenatal exposure, low birth weight, cesarean delivery, maternal medication and nutrition status, pollution, and gut flora.[2,13-19] Predominantly, it is believed that ASD is associated with genetic factors that alter brain development through neural connectivity issues and cause restricted interests and repetitive behaviors.[20] Therefore, interactions between genes and epigenetic influencers can affect specific molecular and cellular pathways associated with ASD. These pathways include gene transcription, mRNA translation, synaptic signaling, abnormal post-translational modifications, and immune and inflammatory aspects.[2] Several genetic variations linked to ASD have been identified and annotated using published scientific literature and whole-exome sequencing (WES).[21] Using the WES approach instead of whole-genome sequencing (WGS) has several advantages. One of them is that it produces a smaller number of results which makes the analysis less challenging. However, while this approach helps us create a list of potential deleterious variants, further research including association and functional studies is needed to determine with more certainty which variants are causing the disease.[22] In 2021, a research team compared the diagnostic yield obtained from different genetic approaches (chromosomal microarrays (CMA), fragile X mental retardation protein (FMR1), and WES) in 343 ASD patients. Compared to CMA or FMR1 testing, WES had shown a statistically significant improvement in diagnostic yield for ASD, making it an appropriate first-tier genetic test to be used. WES had been found to be highly effective in identifying causative variants in patients with ASD. In fact, WES had been found to identify 14.6% of causative variants, while the diagnostic yield of CMA and FMR1 testing was only 2.9% and 0.9%, respectively. About 75% (33/44) of the characterized patients were diagnosed using WES, indicating its superiority over CMA and FMR1 testing. A recent meta-analysis investigated the genetic diagnostic yields of WES and CMA in patients with global developmental delay, ID, and/or ASD. As a result of this analysis, the authors propose a diagnostic algorithm that begins with WES when evaluating unexplained neurodevelopmental disorders.[23]
A previous study was conducted to analyze the exome of 19 trios with ASD from Saudi families by using WES. The aim was to identify de novo or rare inherited coding variants that could have potential clinical relevance. As a result, variants in 15 ASD candidate genes were detected, including 5 genes (GLT8D1, HTATSF1, OR6C65, ITIH6, and DDX26B) that have not been previously reported in any human condition. The remaining identified variants occurred in genes previously linked to ASD or other neurological disorders. The study’s findings were consistent with previous studies, indicating that most of the genes implicated were enriched for biological processes related to neuronal function.[22]
In addition, several studies have been conducted to identify shared pathways and candidate genes for epilepsy or autism using network-based approaches. Generally, these studies use shared biological pathways/processes, protein-protein interaction, co-expression, and other networks to identify relationships between epilepsy or ASD associated genes from sequencing data or selected databases. However, few of these studies have studied ASD and epilepsy in the context of one another.[24] A recent study published in 2020 recommended the use of WES and CMA in the clinical evaluation of children with these conditions. By incorporating these techniques into clinical practice, we can facilitate more accurate diagnoses and treatment plans.[25]
This study aimed to assess the molecular diagnostic yield of DNA samples taken from children with autism and epilepsy and discover their influence on brain development and behavior through WES analysis.
Materials and Methods
Participants
This study was approved by the research and studies department committee-Jeddah Health Affairs (ethical approval no. H-02-J-002). Consent to participate in this study was obtained from the legal guardians of all patients. Only two Saudi families with one male child affected with autism and epilepsy (Trio’s analysis) agreed to participate in this study [Figure 1]. The mean age of the children was 6 years. The children were diagnosed with ASD, ADHD, and epilepsy at the age of three. Recruitment was done through the psychiatry department at East Jeddah Hospital, and the children were assessed by a trained medical team (a pediatric psychiatrist and neurologist) and diagnosed according to the diagnostic and statistical manual of mental disorders (DSM-V) criteria.
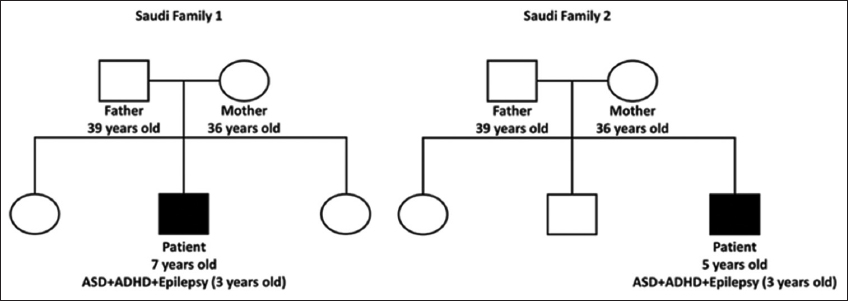
- The pedigrees of the two participant Saudi families. Squares denote males; circles denote females; solid symbols denote affected children. The children were diagnosed with ASD, ADHD, and epilepsy at the age of three. ADHD: Attention-deficit/hyperactivity disorder, ASD: Autism spectrum disorder
DNA extraction
Venous blood samples from parents and affected children were obtained for genomic DNA (gDNA) extraction. DNA extraction was performed in the Center of Excellence in Genomic Medicine Research at King Fahd Medical Research Center, Jeddah, Saudi Arabia. A DNA extraction kit (Qiagen, Hilden, Germany) was used to extract DNA from whole blood samples following the manufacturer’s instructions.
Determination of gDNA concentration
The concentration of the extracted DNA was measured using Qubit® 3.0 Fluorometer (Thermo Fisher Scientific, Catalog no. Q33216, Waltham, MA, USA) using the Qubit quickly and specifically™ dsDNA HS Assay Kit (Thermo Fisher Scientific, Catalog No. Q32851, Waltham, MA, USA).
WES and library generation
WES was selected to uncover the genetic composition of children and their parents. The WES and data analysis for affected children and their parents were carried out by the Bioscience Core Lab at King Abdullah University of Science and Technology (KAUST), Thuwal, Saudi Arabia. RNA/protein contamination was verified by electrophoresis in 1% agarose. To sequence the exotic regions of the samples, Nextera DNA Exome kit (Illumina, Catalog no. 20020616, San Diego, CA, USA) was utilized using 50 ng of gDNA per sample.
Library quantity and quality check (QC)
The concentration of the libraries was measured with Qubit, and the size of the same libraries was measured through 2100 Bioanalyzer instrument (Agilent Technologies, Catalog no. G2939BA, Santa Clara, California, USA) using a High Sensitivity DNA kit (Agilent Technologies, Catalog no. 5067-4626, Santa Clara, California, USA). The libraries were pooled and then hybridized with the probes. Two pools were created: pool 1 included samples 1–3 (first family), and pool 2 included samples 4–6 (second family). After capture and amplification of the libraries, the quality of those was tested again on the Bioanalyzer.
Library quantity assessment
Based on the Qubit quantitation and library size measured through the Bioanalyzer, libraries were pooled equimolarly. The quantitation of the resulting pool was performed using the KAPA Library Quantification kit (Roche, Catalog no. KK4933, Basel, Switzerland) following the manufacturer’s instructions. The final concentration of the pooled libraries was 17nM.
NovaSeq run
Samples were loaded on an Illumina NovaSeq 6000 (S Prime (SP) Flow Cell) SP instrument (Illumina, California, USA) running the 2 × 150 bp paired-end sequencing protocol. The library was diluted to 2pM for final loading and 1% PhiX was added to the pool. For sequencing runs, PhiX was used as a calibration and quality control. PhiX is a nontailed, icosahedral bacteriophage with single-stranded DNA. It contains a small genome of 5386 nucleotides and was Fred Sanger’s first DNA genome to be sequenced. PhiX had been used as a control for Illumina sequencing runs due to its short, well-defined genomic sequence. Illumina advises utilizing PhiX at a low concentration of 1% for most of its library preparations.[26]
Data analysis
The data analysis was conducted by the Biosciences Core Lab at KAUST. The applied statistics were calculated using the Illumina software, NovaSeq control software (version 1.7.0.13), and real-time analysis software (version 3.4.4). The software used for this analysis is part of the following pipeline: bcbio-nextgen (version 1.20.0).
Initial data QC was performed by fast QC (in 00_data and also combined in 01_qc as MultiQC) followed by Mapping through BWA-MEM (BWA). Second data QC (combined in 01_qc as MultiQC), variant calling, and annotation were performed. Family relationship check was done using Genome Analysis Toolkit (GATK version 4.1.6.0)[27] integrated in the bcbio-nextgen (version 1.20.0) pipeline followed by custom filtering using the Gemini tool (version 0.30.2).
Variant’s filtration
The large number of genome variants was filtered using criterions from the Gemini tool.[28] Filtration of less reliable variants was first done by GATK’s hard filtration against VCF files of each family. Quality filter suggested by GATK developers and minimum depth (5 for single nucleotide variant (SNV) and 10 for Indel) was applied. Variants passed the first filter were further filtered by trio information. Impact class was predicted and variants having low impact were ignored. Variants which have a high frequency from databases (higher than 10% in any population) were discarded. Only variants whose known frequencies were low (<10% in any population) were selected. We looked for autism-related variants in genes that were registered in the Simons Foundation Autism Research Initiative (SFARI) database, and the filtration by mode of inheritance (de novo) was determined.
Results
DNA concentration
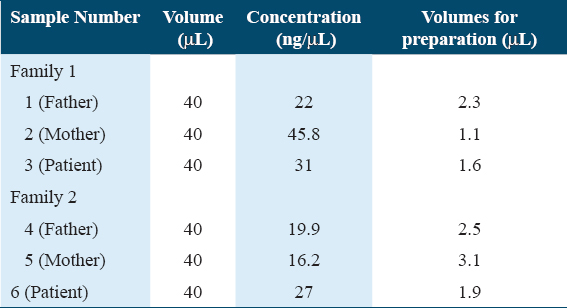
The concentration of the gDNA used in building the libraries is shown in [Table 1].
Library quantity and QC
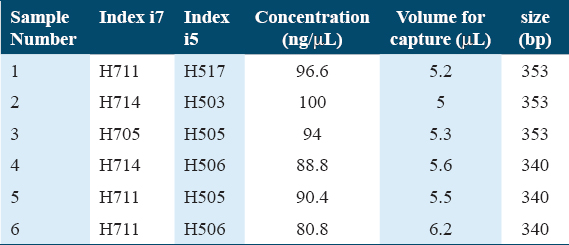
The concentration of the libraries measured with Qubit together with the size of the same libraries measured through the 2100 Bioanalyzer was calculated. The adapter ID that was assigned to each sample library was also reported with Index I showing the traces of each sample library run on the Bioanalyzer. In addition, all libraries show the expected size [Table 2].
Pre-processing and QC
The initial data QC step (fast QC) returned good sequencing quality score distributions and no concerns regarding metrics. Also, family member relationship was checked using variant information computed by Genomic Analysis ToolKit (GATK version 4.1.6.0). Both families showed no contradiction between the datasets and the family trees.
Identification of candidate genetic variants
To interpret phenomenological analysis for screening and annotation, GATK 4 was used to search for small variants (SNP + Indel). The consortium of GATK 4 suggested a quality filter to remove unreliable variants (i.e., wrong predictions). However, each family has numerous variants different from the human reference (hg19). For this reason, a first-round analysis using SNVs was performed and checked to eliminate any errors in indel calls. Furthermore, the current study focused on two scenarios: de novo mutation and autosomal recessive patterns only, to shorten the number of variations. From the given family trees, other types of inheritance, such as autosomal dominant or X-linked cases, were ignored. Family 1 showed 42199 SNVs with 4826 indel calls, while Family 2 showed 46289 SNVs with 5250 indel calls.
De novo mutations in offspring
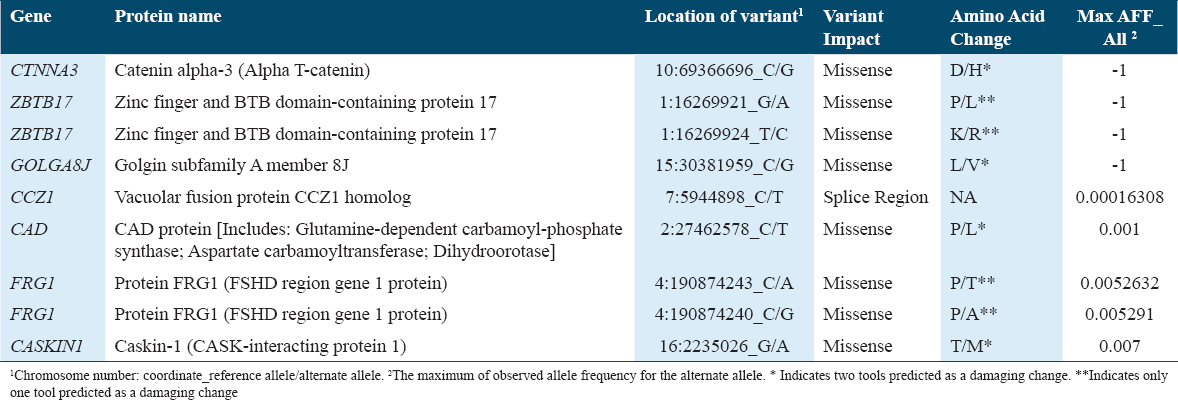
De novo mutations in offspring were annotated using multiple databases such as SFARI Gene, genome aggregation database (gnomAD), and Ensembl Variant Effect Predictor. Variant’s annotations of each gene were reported in Table 3 for family 1 and in Table 4 for family 2.

Discussion
In this study, WES was performed on two Saudi families with one affected child, each presenting with both autism and epilepsy. De novo variations were detected in seven genes (CAD, CTNNA3, ZBTB17, GOLGA8J, CCZ1, FRG1, and CASKIN1) in family 1’s child and four genes (growth differentiation factor-7 [GDF7], NBPF10, DUX4L4, ZNF681) in family 2’s child. Not all of the genes with presumed causative mutations identified here were previously reported in ASD studies.
The CAD gene, located on chromosome 2 (2p23.3), encodes a multifunctional enzyme consisting of carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase,[29] hence the acronym CAD. These enzymes catalyze the first three steps in de novo synthesis of pyrimidine nucleotide, which are of vital importance to DNA, RNA, and metabolites of many cellular processes such as glycosylation and phospholipid synthesis.[30] The CAD gene has been shown to be associated with epilepsy[31] and its mutation causes inborn errors of metabolism and early-infantile epileptic encephalopathy. CAD protein catalyzes the first three steps of the de novo uridine 5’-monophosphate (UMP) biosynthesis. In CAD-deficient patients, oral supplementation with uridine or UMP controlled seizures, and improved development. Growing evidence shows that uridine supplementation can also help patients who suffer deficiencies happening in the early steps of pyrimidine synthesis, including those with CAD mutations.[32-34] In the central nervous system, pyrimidine metabolism defects are thought to cause pathology through pathophysiology cellular mechanisms that are still largely unknown. Successful treatment of epileptic activity may significantly improve cognitive impairment. Genetic testing is the only way to diagnose CAD deficiency due to the lack of biomarkers.[32] Therefore, WES was conducted in this study to predict all the genetic defects the patients could be carrying. In addition, it is crucial to develop thorough, commercially available epilepsy gene panels to screen this population and prevent the long-term effects of inborn errors of metabolism, particularly in the context of the growing number of reported cases of epilepsy.[33]
The Catenin Alpha 3 gene (CTNNA3) is a cell adhesion-encoding protein that has been implicated previously in ASD and other neurodevelopmental disorders.[35-37] Based on western blot analysis, mouse CTNNA3 is highly expressed in the cortex and hippocampus, suggesting it has a neuronal role in early development. Therefore, further studies are needed to investigate the possible role of CTNNA3 in synapse adhesion.[38]
Calcium/calmodulin-dependent serine protein kinase (CASK)-interacting protein 1 or Caskin1 gene was detected as a missense variant. It acts as an adaptor protein regulating cortical actin filaments,[39] plays a role in infantile myoclonic epilepsy,[40] and has been identified as a novel gene associated with the ASD-like phenotype.[41]
Another missense variant gene detected was Golgin subfamily A member 8J (GOLGA8J), which encodes a protein related to Golgi function. A study has shown that a rare genic deletion in the GOLGA8J gene was found in 19% of patients with common forms of genetic epilepsy, which could contribute to brain disorders.[42] Another study also indicated its association with ASD, epilepsy, and mild intellectual disability due to clinically significant copy-number variations.[43]
Two more missense variants were also accounted for the ZBTB17 (MIZ1) gene and FRG1gene. The MIZ1 is a transcription factor with a POZ domain and multiple zinc fingers[44] that are vital for early embryonic development.[45] It functions as a transcription activator when binding to gene promoters,[45] but also can act as a suppressor of many genes such as p15Ink4b, p21Cip1, and Pcdh10 if it was in a complex with the Myc transcription factor.[46-49] The other gene is FRG1, which encodes a protein with an important role in mRNA binding, and muscle development.[50] The down-regulation and/or reduced expression of this gene has been shown to increase the progression of many cancers including breast cancer,[51] prostate cancer[52] as well as angiogenesis.[53] FSHD region gene 1 (FRG1) is a leading candidate for a gene that, if misexpressed, causes facioscapulohumeral muscular dystrophy (FSHD). The expression of FRG1 has been detected in all human tissues studied, including the embryonic brain, placenta, and muscle, suggesting that it has functions outside the muscles.[54] FSHD patients may also present with mental retardation and epilepsy, in addition to the typical features of face, upper arm, and shoulder girdle involvement.[55]
On another note, the CCZ1 gene was the only one detected with a splice region variant. It acts in concert with MON1 to form the MON1-CCZ1 complex to aid the activation of RAB7, a member of the RAS oncogene family and known to regulate the late endosomal/lysosomal network.[56,57] A study has shown that a rare variant in the CCZ1 gene is present in individuals with psychosis and ASD,[58] and another confirmed CCZ1 as a rare and novel candidate gene with a loss variant in ASD cohort only in comparison to the control.[59] These studies support the detection of CCZ1 in this study and suggest that unique and rare variants may be responsible for the occurrence of co-morbid epilepsy and ASD.
The detection of de novo variation in the neuroblastoma breakpoint family member 10 gene (NBPF10) gave a similar interpretation of the results as the other genes in the study. Mutations and SNVs of the NBPF10 gene have been associated with many cancers and syndromes including Mayer–Rokitansky–Küster–Hauser syndrome,[60] hepatocellular carcinoma,[61] formalin-fixed prostate cancer,[62] micropapillary breast carcinoma,[63] and growth hormone deficiency in adults with pituitary stalk interruption syndrome.[64]
In addition, a missense variant of double homeobox protein 4-like protein 4 (DUX4L4) was identified in this study. It is a pseudogene involved in regulation of transcription by RNA polymerase II and its mutation has been associated with B-cell acute lymphoblastic leukemia,[65] and may cause muscular dystrophy in general and FSHD in particular.[66]
Although zinc finger protein 681 (ZNF681) gene was detected with stop-gained variant, it shares similar characteristics and functions with other genes in the study (i.e., it is annotated to have a role in transcription regulation by RNA polymerase II[67] like DUX4L4, and contain a zinc finger domain like ZBTB17). A study using WES revealed that there are 30% common missense and loss-of-function variants shared between metaplastic breast carcinoma and ZNF681.[68]
Finally, the GDF7 gene encodes a protein belonging to a superfamily with roles in the recruitment and activation of SMAD family transcription factors, thus, critically regulating cell development and growth, as well as having growth factor activity properties. In 2020, GDF7 belonged to a list of 11 genes labeled as a risk prognosis model identified to predict survival of patients with endometrial cancer,[69] and later on in 2022, a study showed how it promotes multiple-lineage differentiation in tenogenic cultures of mesenchymal stem cells, as well as stimulates the expression of osteoblastic and adipocytic genes. A study in mice found that GDF7 prevents lipopolysaccharide-induced inflammatory response, oxidative stress, and acute lung injury.[70]
It is believed that the prevalence of ASD is similar across different racial and ethnic groups. However, there is a difference in the average age at which children from different ethnicities are diagnosed with ASD. In the United States, children of African American, Hispanic, and Asian descent are more likely to receive a diagnosis at a later age than Caucasian children. This delay in diagnosis is also observed in some European and Asian countries, though additional research is needed to understand this pattern fully. In addition, studies have shown that there are gender differences in the diagnosis of ASD. Research indicates that ASD is more prevalent among males than females.[71] This is also supported by the findings of the current study, as both families had one male child with ASD and normal female siblings.
Conclusion
The current study identified several genes that were previously not linked to ASD. This discovery opens up new avenues for research and could potentially lead to breakthroughs in our understanding and treatment of ASD. Our findings uncovered several unique genes that are likely to have a role in ASD, as well as providing additional evidence for the function of known ASD genes. These findings suggest two new findings related to ASD. The first is the high amount of genetic heterogeneity, and the second is the fact that the genetic etiology of ASD and other neuropsychiatric and neurodevelopmental illnesses, particularly epilepsy, have a lot in common. According to the results obtained from this study, we concluded that WES can be useful in the evaluation of children with ASD and epilepsy, and should be considered in clinical practice. Furthermore, future functional studies are needed to determine the specific roles of genes and variants in each patient and this knowledge should be translated into knowledge of the molecular basis of ASD that can help patients better understand their condition and then begin developing more effective treatments. Further analysis with a larger sample of ASD and/or ASD-epilepsy patients will aid in a better understanding of the genetic and variants mapping and provide possible new insights.
Limitations
This study has some potential limitations, such as a small sample size due to limited cooperation from families and most of the patients’ parents declined genetic testing on their children. In addition, we self-funded this project, and the cost of carrying a large number of samples was quite expensive. Although this study included a small sample size, it is still valuable as it provides a rich dataset that uncovered new ASD candidate genes.
Ethical approval and patient’s consent
This study was approved by the research and studies department committee-Jeddah Health Affairs (ethical approval no. H-02-J-002). Consent to participate in this study was obtained from the legal guardians of all patients.
Funding Statement
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Competing Interest
All the authors declared that there was no conflict of interest. All authors declared that the work is original and does not infringe copyright or other parties’ property rights. All authors have read and approved this submission and have given appropriate credit to everyone who participated in this work.
Availability of Data
The data supporting the findings of this study are available within the article. More detailed data used to support the findings of the current study are available from the corresponding author on reasonable request.
Authors’ Contribution
AAA conceived and designed the project, AAB, AS, AHA, and RAA acquired resources and performed the statistical analysis, MME optimized the methodology, recruitment, and diagnosis of patients, AAA and AAB acquired, analyzed, and interpreted the data, MHA project administrator, AAB wrote the first draft of the paper, AAA and MMA wrote the manuscript.
Consent for Publication
The authors jointly approved the publication of this manuscript.
References
- Autism spectrum disorder:Neurodevelopmental risk factors, biological mechanism, and precision therapy. Int J Mol Sci. 2023;24:1819.
- [Google Scholar]
- Overview and introduction to autism spectrum disorder (ASD) Adv Neurobiol. 2020;24:3-42.
- [Google Scholar]
- A delayed diagnosis of autism spectrum disorder in the setting of complex attention deficit hyperactivity disorder. Cureus. 2022;14:e25825.
- [Google Scholar]
- Vitamin A status is more commonly associated with symptoms and neurodevelopment in boys with autism Spectrum disorders-a multicenter study in China. Front Nutr. 2022;9:851980.
- [Google Scholar]
- The global prevalence of autism spectrum disorder:A comprehensive systematic review and meta-analysis. Ital J Pediatr. 2022;48:112.
- [Google Scholar]
- Prevalence of autism spectrum disorder in Asia:A systematic review and meta-analysis. Psychiatry Res. 2020;284:112679.
- [Google Scholar]
- Systemic review of the epidemiology of autism in Arab Gulf countries. Neurosciences (Riyadh). 2014;19:291-6.
- [Google Scholar]
- Prevalence and clinical characteristics of autism spectrum disorders in school-age children in Taif- KSA. Int J Med Sci Public Health. 2013;2:556.
- [Google Scholar]
- Effect of autism on parental quality of life in Arar city, Saudi Arabia. J Family Community Med. 2020;27:15-22.
- [Google Scholar]
- Prevalence and characteristics of autistic children attending autism centres in 2 major cities in Saudi Arabia:A cross-sectional study. Saudi Med J. 2021;42:419-27.
- [Google Scholar]
- Prevalence of autism spectrum disorder among Saudi children between 2 and 4 years old in Riyadh. Asian J Psychiatr. 2022;71:103054.
- [Google Scholar]
- Pathways to a diagnosis of autism spectrum disorder in Germany:A survey of parents. Child Adolesc Psychiatry Ment Health. 2019;13:16.
- [Google Scholar]
- Autism spectrum disorder:Definition, epidemiology, causes, and clinical evaluation. Transl Pediatr. 2020;9:S55-65.
- [Google Scholar]
- The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell Mol Life Sci. 2019;76:1275-97.
- [Google Scholar]
- Risk for autism spectrum disorders according to period of prenatal antidepressant exposure:A systematic review and meta-analysis. JAMA Pediatr. 2017;171:555-6.
- [Google Scholar]
- Maternal vitamin D deficiency and the risk of autism spectrum disorders:Population-based study. BJPsych Open. 2016;2:170-2.
- [Google Scholar]
- Neurodevelopmental disorders and prenatal residential proximity to agricultural pesticides:The CHARGE study. Environ Health Perspect. 2014;122:1103-9.
- [Google Scholar]
- Prevalence of autism spectrum disorder in preterm infants:A meta-analysis. Pediatrics. 2018;142:e20180134.
- [Google Scholar]
- Neuroimaging in autism spectrum disorder:Brain structure and function across the lifespan. Lancet Neurol. 2015;14:1121-34.
- [Google Scholar]
- AutDB:A gene reference resource for autism research. Nucleic Acids Res. 2009;37:D832-6.
- [Google Scholar]
- Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder:A trio study from Saudi families. Sci Rep. 2017;7:5679.
- [Google Scholar]
- Towards a change in the diagnostic algorithm of autism spectrum disorders:Evidence supporting whole exome sequencing as a first-tier test. Genes. 2021;12:560.
- [Google Scholar]
- Ravdin LD, Katzen HL, eds. Epilepsy and aging BT- Handbook on the Neuropsychology of Aging and Dementia. Cham: Springer International Publishing; 2019. p. :401-25.
- Clinical and genetic profile of autism spectrum disorder-epilepsy (ASD-E) phenotype:Two sides of the same coin! Clin EEG Neurosci. 2020;51:390-8.
- [Google Scholar]
- Large-scale contamination of microbial isolate genomes by Illumina PhiX control. Stand Genomic Sci. 2015;10:18.
- [Google Scholar]
- Genomics in the Cloud:Using Docker, GATK, and WDL in Terra (1st ed). United States: O'Reilly Media; 2020.
- GEMINI:Integrative exploration of genetic variation and genome annotations. PLoS Comput Biol. 2013;9:e1003153.
- [Google Scholar]
- Deciphering CAD:Structure and function of a mega-enzymatic pyrimidine factory in health and disease. Protein Sci. 2021;30:1995-2008.
- [Google Scholar]
- CAD mutations and uridine-responsive epileptic encephalopathy. Brain. 2017;140:279-86.
- [Google Scholar]
- Expanding the clinical and genetic spectrum of CAD deficiency:An epileptic encephalopathy treatable with uridine supplementation. Genet Med. 2020;22:1589-97.
- [Google Scholar]
- Triacetyluridine treats epileptic encephalopathy from CAD mutations:A case report and review. Ann Clin Transl Neurol. 2021;8:284-7.
- [Google Scholar]
- CAD gene and early infantile epileptic encephalopathy-50;three Iranian deceased patients and a novel mutation:Case report. BMC Pediatr. 2022;22:125.
- [Google Scholar]
- Investigating the effects of copy number variants on reading and language performance. J Neurodev Disord. 2016;8:17.
- [Google Scholar]
- Common genetic variants on 5p14.1 associate with autism spectrum disorders. Nature. 2009;459:528-33.
- [Google Scholar]
- ACTNNA3 compound heterozygous deletion implicates a role for αT-catenin in susceptibility to autism spectrum disorder. J Neurodev Disord. 2014;6:17.
- [Google Scholar]
- Genetic association of CTNNA3 with late-onset Alzheimer's disease in females. Hum Mol Genet. 2007;16:2854-69.
- [Google Scholar]
- CASK participates in alternative tripartite complexes in which mint 1 competes for binding with Caskin 1, a novel CASK-binding protein. J Neurosci. 2002;22:4264-73.
- [Google Scholar]
- Prediction of human disease genes by human-mouse conserved coexpression analysis. PLoS Comput Biol. 2008;4:e1000043.
- [Google Scholar]
- Hippocampal transcriptomic and proteomic alterations in the BTBR mouse model of autism spectrum disorder. Front Physiol. 2015;6:324.
- [Google Scholar]
- Rare gene deletions in genetic generalized and Rolandic epilepsies. PLoS One. 2018;13:e0202022.
- [Google Scholar]
- Comparative analyses of copy-number variation in autism spectrum disorder and schizophrenia reveal etiological overlap and biological insights. Cell Rep. 2018;24:2838-56.
- [Google Scholar]
- The POZ domain:A conserved protein-protein interaction motif. Genes Dev. 1994;8:1664-77.
- [Google Scholar]
- Miz1 is required for early embryonic development during gastrulation. Mol Cell Biol. 2003;23:7648-57.
- [Google Scholar]
- Miz1 promotes KRAS-driven lung tumorigenesis by repressing the protocadherin Pcdh10. Cancer Lett. 2023;555:216025.
- [Google Scholar]
- The zinc finger protein Miz1 suppresses liver tumorigenesis by restricting hepatocyte-driven macrophage activation and inflammation. Immunity. 2021;54:1168-85.e8.
- [Google Scholar]
- Myc-Miz1 signaling promotes self-renewal of leukemia stem cells by repressing Cebpα and Cebpδ. Blood. 2020;135:1133-45.
- [Google Scholar]
- Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. 2001;3:392-9.
- [Google Scholar]
- FRG1 is a direct transcriptional regulator of nonsense-mediated mRNA decay genes. Genomics. 2023;115:110539.
- [Google Scholar]
- Reduced expression of FRG1 facilitates breast cancer progression via GM-CSF/MEK-ERK axis by abating FRG1 mediated transcriptional repression of GM-CSF. Cell Death Discov. 2022;8:442.
- [Google Scholar]
- Reduced FRG1 expression promotes prostate cancer progression and affects prostate cancer cell migration and invasion. BMC Cancer. 2019;19:346.
- [Google Scholar]
- Reduced FRG1 expression promotes angiogenesis via activation of the FGF2-mediated ERK/AKT pathway. FEBS Open Bio. 2023;13:804-17.
- [Google Scholar]
- FSHD region gene 1 (FRG1) is crucial for angiogenesis linking FRG1 to facioscapulohumeral muscular dystrophy-associated vasculopathy. Dis Model Mech. 2009;2:267-74.
- [Google Scholar]
- Facioscapulohumeral muscular dystrophy with severe mental retardation and epilepsy. Brain Dev. 2007;29:231-3.
- [Google Scholar]
- RAB7 activity is required for the regulation of mitophagy in oocyte meiosis and oocyte quality control during ovarian aging. Autophagy. 2022;18:643-60.
- [Google Scholar]
- Structure of the Mon1-Ccz1 complex reveals molecular basis of membrane binding for Rab7 activation. Proc Natl Acad Sci U S A. 2022;119:e2121494119.
- [Google Scholar]
- Copy number variants in people with autism spectrum disorders and co-morbid psychosis. Eur J Med Genet. 2018;61:230-4.
- [Google Scholar]
- Rare copy number variations in a Chinese cohort of autism spectrum disorder. Front Genet. 2018;9:665.
- [Google Scholar]
- Detection of de novo genetic variants in Mayer-Rokitansky-Küster-Hauser syndrome by whole genome sequencing. Eur J Obstet Gynecol Reprod Biol X. 2019;4:100089.
- [Google Scholar]
- Somatic mutation profiles revealed by next generation sequencing (NGS) in 39 Chinese hepatocellular carcinoma patients. Front Mol Biosci. 2021;8:800679.
- [Google Scholar]
- Mutation detection in formalin-fixed prostate cancer biopsies taken at the time of diagnosis using next-generation DNA sequencing. J Clin Pathol. 2015;68:212-7.
- [Google Scholar]
- Characterization of the genomic features and expressed fusion genes in micropapillary carcinomas of the breast. J Pathol. 2014;232:553-65.
- [Google Scholar]
- Normal height and novel mutations in growth hormone deficiency adults with pituitary stalk interruption syndrome. Neuro Endocrinol Lett. 2019;40:299-304.
- [Google Scholar]
- Long-read sequencing unveils IGH-DUX4 translocation into the silenced IGH allele in B-cell acute lymphoblastic leukemia. Nat Commun. 2019;10:2789.
- [Google Scholar]
- Predicting novel genomic regions linked to genetic disorders using GWAS and chromosome conformation data - a case study of schizophrenia. Sci Rep. 2019;9:17940.
- [Google Scholar]
- Phylogenetic-based propagation of functional annotations within the Gene Ontology consortium. Brief Bioinform. 2011;12:449-62.
- [Google Scholar]
- Quantitative proteomic landscape of metaplastic breast carcinoma pathological subtypes and their relationship to triple-negative tumors. Nat Commun. 2020;11:1723.
- [Google Scholar]
- The role of N6-methyladenosine methylation in the progression of endometrial cancer. Cancer Biother Radiopharm. 2020;37:737-49.
- [Google Scholar]
- Growth differentiation factor 7 prevents sepsis-induced acute lung injury in mice. Evid Based Complement Alternat Med. 2022;2022:3676444.
- [Google Scholar]
- The diagnosis of autism:From Kanner to DSM-III to DSM-5 and beyond. J Autism Dev Disord. 2021;51:4253-70.
- [Google Scholar]




