
Translate this page into:
Nephrotic syndrome: Pretibial epidermolysis bullosa in a patient with CD151 tetraspanin defect: A case report
Address for correspondence: Raniah S. Alotibi, Department of Clinical Laboratory Sciences, College of Applied Medical Sciences, King Saud Bin Abdulaziz University for Health Sciences, Riyadh, Saudi Arabia. Phone: +966-1144299999. E-mail: raniao@ksau-hs.edu.sa
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Qassim Uninversity and was migrated to Scientific Scholar after the change of Publisher.
Abstract
ABSTRACT
Nephrotic syndrome (NS)-epidermolysis bullosa (EB) sensorineural deafness syndrome is an autosomal recessive rare genetic disease caused by a CD151 gene homozygous mutation on chromosome 11p15.5. In this report, we discuss a rare case related to a Saudi patient with genetic syndrome who presented with NS and EB. Whole genome sequencing results indicated a homozygous pathogenic variant identified in the CD151 gene (c.493C>T p.(Arg165*), which was consistent with a genetic diagnosis of autosomal recessive nephropathy with pretibial EB and deafness syndrome. The findings emphasize that even a single genotype can result in variable phenotypic expression, necessitating the assessment of the pleiotropic effects of the disease on the patient, which can range from severe to mild. This case report adds to the literature by highlighting the considerable phenotypic variation that can be present in patients with the CD151 mutation.
Keywords
CD151 gene
genetic disease
nephrotic syndrome
pathogenic variant

Introduction
Nephrotic syndrome (NS) is a presentation of glomerular disease characterized by proteinuria, hypoalbuminemia, and edema, with or without hyperlipidemia.[1] If untreated, NS can lead to significant complications, such as edema with respiratory compromise, risk of infections, hypercoagulability, and risk of end-stage renal disease (ESRD); hence, early detection and treatment are crucial. NS can be due to either primary or secondary causes.[1] The list of primary causes is endless and includes NS related to genetic mutations.
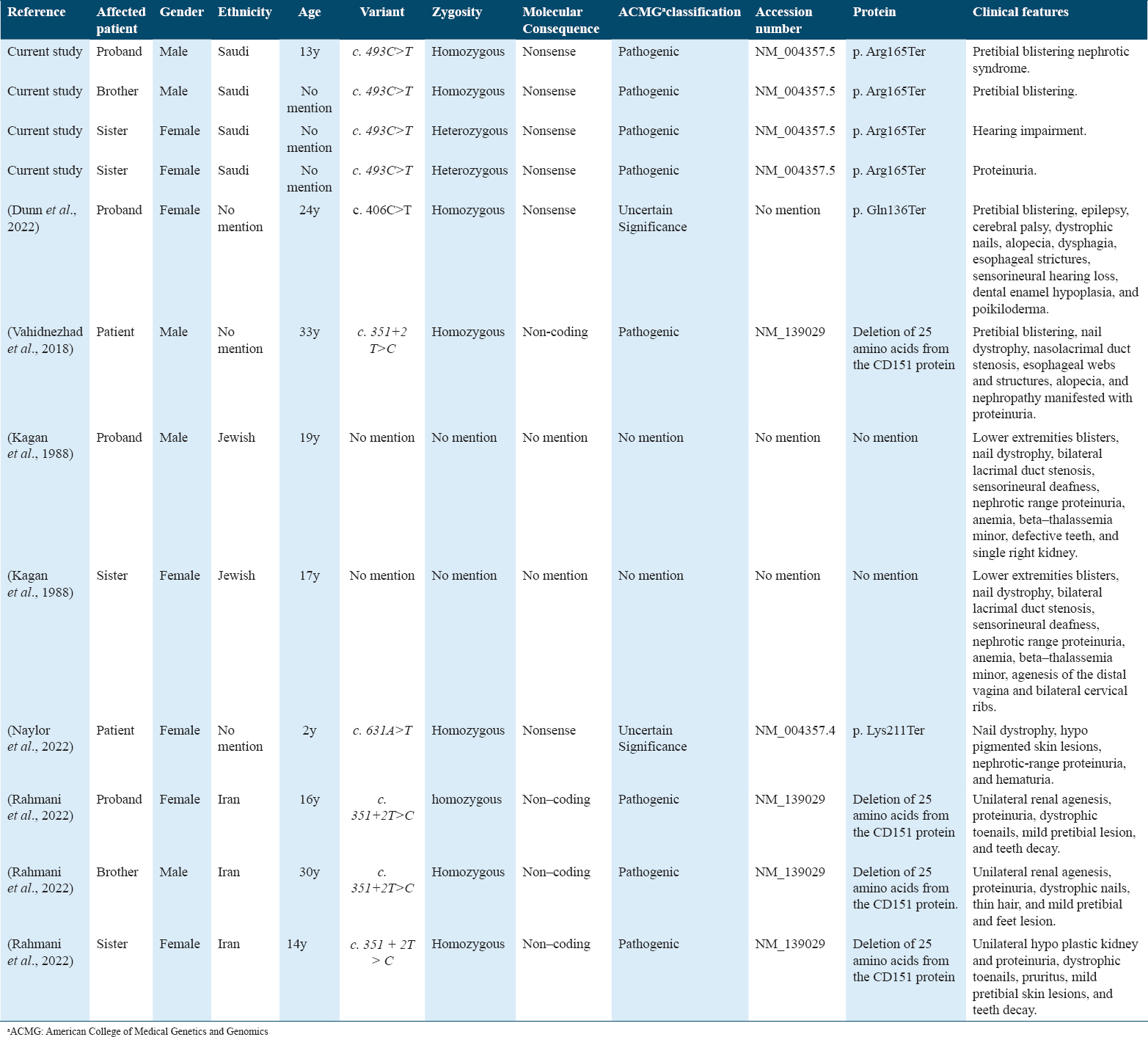
NS-epidermolysis bullosa (EB) sensorineural deafness syndrome is an autosomal recessive genetic disease caused by a CD151 gene homozygous mutation on chromosome 11p15.5.[2] This syndrome is reported to have an adolescent age of onset and is characterized by hereditary nephritis, leading to NS and ESRD, associated with sensorineural hearing loss and pretibial skin blistering [Table 1].[2] Other manifestations have been reported, such as bilateral cervical ribs, unilateral kidneys, dystrophic teeth and nails, distal vaginal agenesis, anemia due to beta-thalassemia minor, and bilateral lacrimal duct stenosis.[2]

The human CD151 gene is located on chromosome 11p15.5 and comprises 8 exons, with exons 2–8 encoding the CD151 polypeptide.[3] CD151 has a wide cell and tissue distribution, including the epithelium, endothelium, muscle, renal glomeruli and proximal and distal tubules, Schwann cells, and dendritic cells, with a single ribonucleic acid species observed in most human adult tissues. CD151 is expressed at high levels in platelets and megakaryocytes.[3] The protein encoded by this gene is a member of the transmembrane 4 superfamily, also known as the tetraspanin family. Tetraspanins are cell surface proteins that play a role in the regulation of cell development, activation, growth, and motility. These proteins mediate signal transduction events that play a role in the regulation of cell development, activation, growth, and motility.[3] CD151 is essential for the normal function of glomerular and tubular basement membranes in the kidney, as well as skin and inner ear functions, and may also play a role in erythropoiesis.[3] In this report, we discuss a rare case related to a Saudi patient with genetic syndrome who presented with NS and EB.
Case Presentation
A 13-year-old boy was brought by his mother to the emergency room in February 2019 with a history of incidental finding of proteinuria 6 months earlier, when the mother had tested his urine using a urine dipstick test at home. At that time (6 months ago), he was taken to a local hospital in the family’s hometown and found to have a total urine protein of 2070 mg/day (56 mg/kg/day); both C3 and C4 levels were normal, and the serum albumin level was normal (34 g/L). A renal ultrasound was conducted and demonstrated a small right kidney measuring 4 cm. No treatment was initiated at that time, and he was brought by his parents to our hospital for further investigations.
The patient was full-term at birth and delivered through spontaneous vaginal delivery. Pregnancy was normal, with no prenatal, perinatal, or postnatal complications. The patient’s medical history was significant due to a previous episode of bullous lesions in the lower limbs, which resolved without medical intervention. The patient’s family consisted of four brothers and two sisters. One sister had resolved proteinuria (having received steroids), another sister had a hearing impairment and low-grade proteinuria, and one brother had experienced recurrent bullous lesions. The parents were first-degree relatives and asymptomatic. At admission, the patient was asymptomatic, vitally stable, and normotensive and had a normal examination (i.e., no dysmorphic features, clinical edema, active skin lesions, nails, or teeth changes were observed). He maintained adequate urine output throughout his admission. The initial blood examination showed the following outcomes: normal blood cell counts with no leukocytosis, anemia, or thrombocytopenia. Serum albumin and total urine protein were slightly low at 32 g/L (normal range: 38–54 g/L) and 55 g/L (normal range: 60–83), respectively. He had normal renal function, his creatinine was within the normal range of 47 μmol/L (normal range: 42–71 μmol/L), and his blood urea nitrogen was 6.3 mmol/L and thus within the normal range (2.5–10.7 mmol/L). The liver enzymes were also normal, and C3 and C4 levels were within normal ranges.
Autoimmune tests were negative for anti-neutrophil cytoplasmic antibodies (ANCA), anti-double-strandeddeoxyribonucleic acid (DNA), and perinuclear ANCA (MPO-ANCA). Serum anti-proteinase 3 antineutrophil cytoplasmic antibody (PR3-ANC) and hepatitis screen were also negative. Initial urine analyses showed a high urine protein, >600 mg/dL, with microscopic hematuria of 12 red blood cells/high power field and 24-h urine protein of 5.32 g/day (normal ≤0.30 g/day). A renal ultrasound was performed and showed an atrophied right kidney measuring 4 cm with compensatory hypertrophy of the left one, which measured 12 cm. A Dimercapto succinic acid renal scan was carried out and showed that the right kidney had limited function: the right kidney had a calculated function of 2% and the left one of 98%. At admission, the patient was given an oral angiotensin-converting enzyme (ACE) inhibitor as an initial treatment for anti-proteinic properties and was discharged with a follow-up plan with both the pediatric nephrology and genetic departments. Figure 1 illustrates his proteinuria trend and shows resolution with relapses, bearing in mind that the patient showed poor compliance with the oral ACE inhibitor.
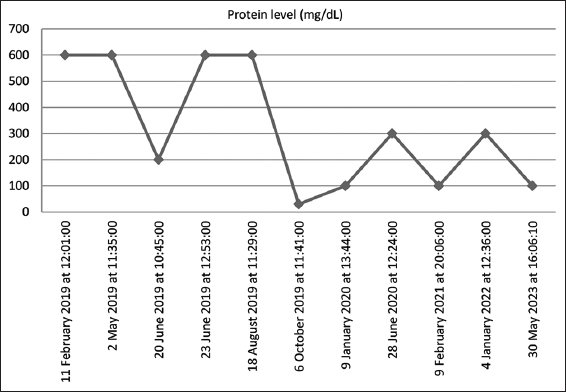
- Trend values of proteinuria since 2019
Whole genome sequencing (WGS) was performed in a commercial College of American Pathologists/Clinical Laboratory Improvement Amendments-accredited laboratory. Illumina HiSeq 4000 sequencers were used for WGS. The average coverage depth for WGS was ~30X. For the extended family analysis, Sanger sequencing was carried out. Several tools were used for the data analysis, including Alamut® Visual (http://www.interactive-biosoftware.com/alamut-visual/), BaseSpace Variant Interpreter (http://variantinterpreter.informatics.illumina.com/home), and Varsome “the Human Genomics Community” (https://varsome.com/). The predicted classification of a variant as pathogenic/likely pathogenic, of uncertain significance, or benign was based on the American College of Medical Genetics and Genomics scoring system.[4] Detailed clinical information on the family according to the human phenotype ontology format was as follows: NS (HP: 0000100), which refers to the constellation of clinical findings that result from severe renal loss of protein, with proteinuria and hypoalbuminemia, edema, and hyperlipidemia; pretibial blistering (HP: 0012221), a type of blistering that affects the skin of the tibial region; hearing impairment (HP: 0000365), a decreased magnitude of the sensory perception of sound; and proteinuria (HP: 0000093), increased levels of protein in the urine.
WGS results indicated a homozygous pathogenic variant identified in the CD151 gene (c.493C>T p.(Arg165*), which was consistent with a genetic diagnosis of autosomal recessive nephropathy with pretibial EB and deafness syndrome. The patient was referred to the genetic team, and segregation analysis was performed for his family, as detailed in Figure 2. A hearing assessment of the patient showed normal middle ear function and hearing levels bilaterally. Hemoglobin electrophoresis was carried out and showed a normal level of hemoglobin component; hence, the result was negative for thalassemia. The patient was discharged in good condition with the resolution of his proteinuria. He was kept on PO Lisinopril with follow-up visits. The patient had relapses of proteinuria that are most likely explained by poor compliance with Lisinopril. His kidney function remained normal throughout his follow-up, as the proteinuria trend is illustrated in Figure 1.
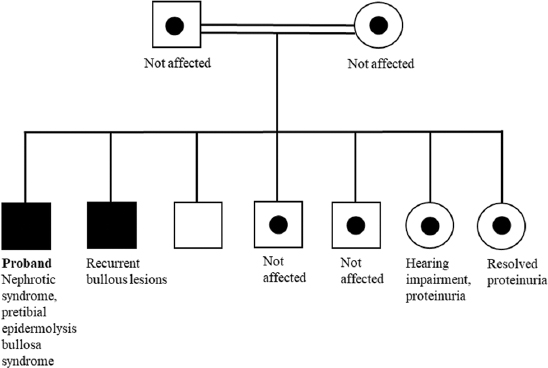
- Family pedigree of the patient
Discussion
Among the CD151-related syndromes, a few cases of nephropathy were noted in the form of proteinuria associated with EB, with highly variable clinical presentation [Table 2]. A 24-year-old woman with EB, nephropathy, epilepsy, and cystic fibrosis was homozygous for an autosomal recessive c.406C>T mutation in exon 6 of the CD151 gene.[5] A 33-year-old man had a homozygous CD151 mutation, leading to aberrant splicing of exon 5 and deletion of 25 amino acids from CD151. This mutation was associated with the clinical diagnosis of Kindler syndrome, presenting as EB, nail dystrophy, nasolacrimal duct stenosis, esophageal webs and structures, alopecia, and nephropathy as proteinuria.[6] In 1988, EB was reported in two siblings in an Oriental Jewish family.[7] Both siblings had hereditary nephritis, pre-tibial EB, and beta-thalassemia minor with nephrotic range proteinuria and ESRD and were on peritoneal dialysis. They also had other features, including nasolacrimal duct stenosis, nail dystrophy, and sensorineural hearing loss.[7] In this study, we describe the cases of four siblings with CD151 mutation c.493C>T who displayed varying phenotypes among themselves and in comparison with previous reports [Table 2]. Hearing impairment and NS were not present in the proband and his brother, who inherited the homozygous version of the mutation. Furthermore, the discrepancy in hearing impairment was remarkable: although both sisters carried heterozygous c.493C>T, one had hearing impairment, and the other had only proteinuria. The other two brothers and parents, who carried heterozygous versions, remained asymptomatic.
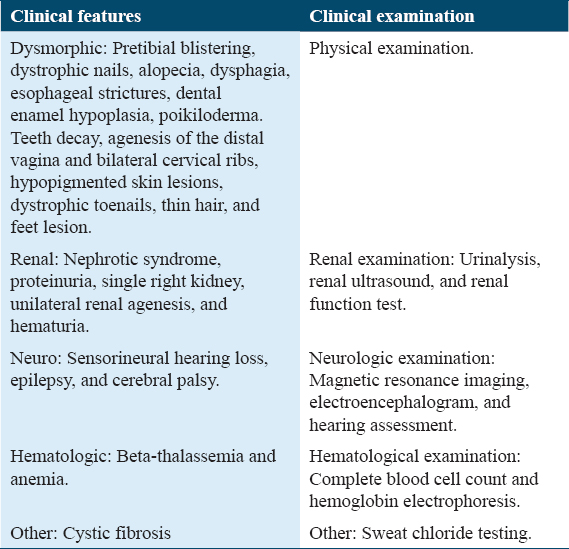
A wide spectrum of phenotypic expression in patients with CD151 mutations may be controlled by epigenetic factors.[8] Of note, epigenetic mechanisms also contribute to the variation among organisms with identical chromosomal DNA sequences, such as in human monozygotic twins, when phenotypic variation occurs in the absence of environmental factors and when environmental factors do not considerably increase the degree of phenotypic variation.[8] Moreover, variations in presentation and severity can be due to the effect of other modifier genes; for example, TSPAN11, another member of the tetraspanin family, has a partial modifying role in CD151 functions.[9] Importantly, clinicians might need to assess the pleiotropic effects of the disease in patients with CD151 mutations and consider all possible clinical features reported previously. Table 3 presents the recommended diagnostic evaluation of patients with CD151 mutations.
This study is a report of a very rare syndrome caused by a c.493C>T mutation in CD151. The findings emphasize that even a single genotype can result in variable phenotypic expression, necessitating the assessment of the pleiotropic effects of the disease on the patient, which can range from severe to mild. This variation in phenotype and severity may be explained by epigenetic factors and other modifier genes. The findings strongly support that the rare genetic variant is pathogenic, with variable severity. Our patient presented with nephrotic range proteinuria, unilateral kidney with nonfunctioning atrophied right kidney, and a history of lower limb bilious lesions. He did not have hearing impairments or other conditions mentioned in previous reports. Notably, as evident from his family history, this gene mutation has variable expressivity and phenotypic expression. He had one sister with isolated steroid-responsive NS (heterozygous), another sister with hearing impairment and low-grade proteinuria (heterozygous), and one brother with recurrent lower limb bullous lesions (homozygous).
A study examined the mechanism of CD151 deficiency leading to kidney failure in CD151-null mice and discovered that the mice developed massive proteinuria caused by focal glomerulosclerosis, disorganization of the glomerular basement membrane, and tubular cystic dilation, but their skin integrity or hearing ability remained unchanged.[10] Moreover, generated podocyte-specific conditional knockout mice for the integrin a3 subunit had renal defects similar to those in the CD151-knockout mice; thus, CD151 was suggested to play a key role in strengthening a3b1-mediated adhesion in podocytes.[10]
Conclusion
The syndrome of NS, EB, and sensorineural deafness secondary to the CD151 mutation is a very rare autosomal recessive disease. Thus, this case report adds to the literature by highlighting the considerable phenotypic variation that can be present in patients with the CD151 mutation. Further reports of similar cases are critical to extending our understanding of the phenotypic spectrum and long-term prognosis of this rare genetic disease.
Author’s Declaration Statement
Availability of data
The data presented in the current case are deposited in the ClinVar database repository, accession numberSCV004012070. SUB13655658 - ClinVar- File Submission Portal (nih.gov).
Patient consent statement
The patient and family have been consented and a written consent form was obtained from patients’ parents.
Funding statement
There is no funding agent for the current research.
Role of the funding source
None.
Competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Author contributions
KA and HA: Conceptualized the idea, collected data, and wrote the first draft of the manuscript. MA and RA: Analyzed data and contributed to the manuscript writing. All authors reviewed and approved the final draft of the manuscript.
References
- Consensus reclassification of inherited epidermolysis bullosa and other disorders with skin fragility. Br J Dermatol. 2020;183:614-27.
- [Google Scholar]
- CD151, the first member of the tetraspanin (TM4) superfamily detected on erythrocytes, is essential for the correct assembly of human basement membranes in kidney and skin. Blood. 2004;104:2217-23.
- [Google Scholar]
- Standards and guidelines for the interpretation of sequence variants:A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405-24.
- [Google Scholar]
- Expanding the spectrum of epidermolysis bullosa simplex:Syndromic epidermolysis bullosa simplex with nephropathy and epilepsy secondary to CD151 tetraspanin defect-a case report and review of the literature. JAAD Case Rep. 2022;23:136-40.
- [Google Scholar]
- Recessive mutation in tetraspanin CD151 causes kindler syndrome-like epidermolysis bullosa with multi-systemic manifestations including nephropathy. Matrix Biol. 2018;66:22-33.
- [Google Scholar]
- Letters to the editor occurrence of hereditary nephritis, prétibial epidermolysis bullosa and beta-thalassemia minor in two siblings with end-stage renal disease. Nephron. 1988;49:331-2.
- [Google Scholar]
- Phenotypic differences in genetically identical organisms:The epigenetic perspective. Hum Mol Genet. 2005;14:R11-8.
- [Google Scholar]
- New report of a different clinical presentation of CD151 splicing mutation (c.351+2T>C):Could TSPAN11 be considered as a potential modifier gene for CD151 ? Mol Syndromol. 2022;13:212-20.
- [Google Scholar]




