
Translate this page into:
RSAD2: An exclusive target protein for Zika virus comparative modeling, characterization, energy minimization and stabilization
Address for correspondence: Dr. Sachidanand Singh, Department of Biotechnology, Sankalchand Patel University, Visnagar - 384 315, Gujarat, India. E-mail: drsachinbioinfo@gmail.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Qassim Uninversity and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Objective:
The major purpose of the present study was to predict the structure of Radical s-adenosyl-L-methionine Domain 2 (RSAD2), the most targeted protein of the Zika virus using comparative modeling, to validate the models that were generated and molecular dynamics (MD) simulations were performed.
Methods:
The secondary structure of RSAD2 was estimated using the Garnier-Osguthorpe-Robson, Self-Optimized Prediction method with Alignment, and Position-Specific Iterative-Blast based secondary structure prediction algorithms. The best of them were preferred based on their DOPE score, then three-dimensional structure identification using SWISS-MODEL and the Protein Homology/Analogy Recognition Engine (Phyre2) server. SAVES 6.0 was used to validate the models, and the preferred model was then energetically stabilized. The model with least energy minimization was used for MD simulations using iMODS.
Results:
The model predicted using SWISS-MODEL was determined as the best among the predicted models. In the Ramachandran plot, there were 238 residues (90.8%) in favored regions, 23 residues (8.8%) in allowed regions, and 1 residue (0.4%) in generously allowed regions. Energy minimization was calculated using Swiss PDB viewer, reporting the SWISS-MODEL with the lowest energy (E = −18439.475 KJ/mol) and it represented a stable structure conformation at three-dimensional level when analyzed by MD simulations.
Conclusion:
A large amount of sequence and structural data is now available, for tertiary protein structure prediction, hence implying a computational approach in all the aspects becomes an opportunistic strategy. The best three-dimensional structure of RSAD2 was built and was confirmed with energy minimization, secondary structure validation and torsional angles stabilization. This modeled protein is predicted to play a role in the development of drugs against Zika virus infection.
Keywords
Comparative modeling
energy minimization
molecular dynamics
protein homology/analogy recognition engine
saves 6.0
SWISS-MODEL

Introduction
Zika virus is an emerging virus reported by the World Health Organization as a serious global biological threat.[1] The Zika virus was initially discovered by Alexander John Haddow (an entomologist). The Zika virus is a mosquito-borne flavivirus that has been related to serious birth problems in newborns, including microcephaly, and is linked to Guillain-Barre syndrome.[2] Radical s-adenosyl-L-methionine Domain 2 (RSAD2) proteins have recently been investigated for their possible role in the Zika virus. The interferon-inducible antiviral protein RSAD2 encoded by this gene belongs to the S-Adenosyl-L-Methionine (SAM) super family of enzymes. RSAD2 is involved in the cellular antiviral response as well as innate immune signaling. In the Zika virus, this protein has been discovered to block both DNA and RNA.[3]
The radical SAM activity of RSAD2(Viperin) reduces the NAD+ -dependent activity of enzymes that control metabolism. It catalyzes the conversion of Cytidine Triphosphate (CTP) to its analogue 3’-deoxy-3’, 4’-didehydro-CTP, which was recently found (ddhctp). This metabolite’s biological function is still unknown. RSAD2 expression decreased the level of NS3 (nonstructural) protein and the stability of the other interacting viral protein, only when NS3 was present. The Zika virus proved susceptible toward RSAD2’s antiviral action. This sensitivity was linked to RSAD2’s ability to degrade NS3 in a proteasome-dependent manner.[4] When NS3 was removed, Zika was entirely rescued, indicating that the viral NS3 is the RSAD2 protein’s particular target. RSAD2, a host protein produced in response to infection, effectively prevents several Flaviviruses from replicating.[5] In the present work, comparative protein structure modeling and its characterization with Ramachandran plot is been performed for the RSAD2. Moreover, energy minimization and molecular dynamics (MD) simulations were performed for the modeled and validated protein for stable confirmations. The outcomes of this study offer a plausible common mechanism, according to which the RSAD2 protein may be further explored at active site and molecular docking level, an alternative strategy for medical research in Zika virus infection.
Materials and Methods
MethodsThere are a series of steps in the comparative modeling of protein. First, the target must be identified, and then the target and template sequences must be aligned. Steric clashes and stability must be discovered, once the template sequence alignment is accomplished [Figure 1].
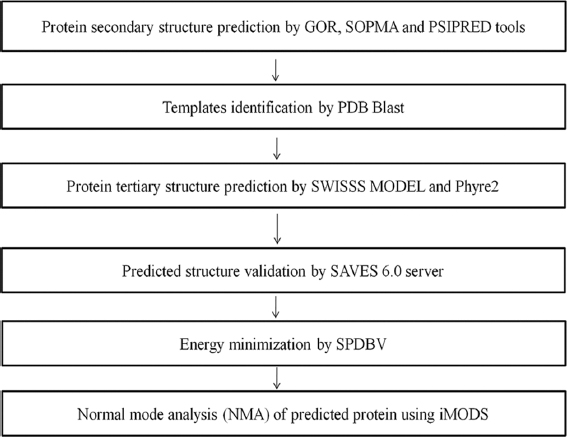
- Molecular modeling Flow chart
Secondary structure prediction of protein
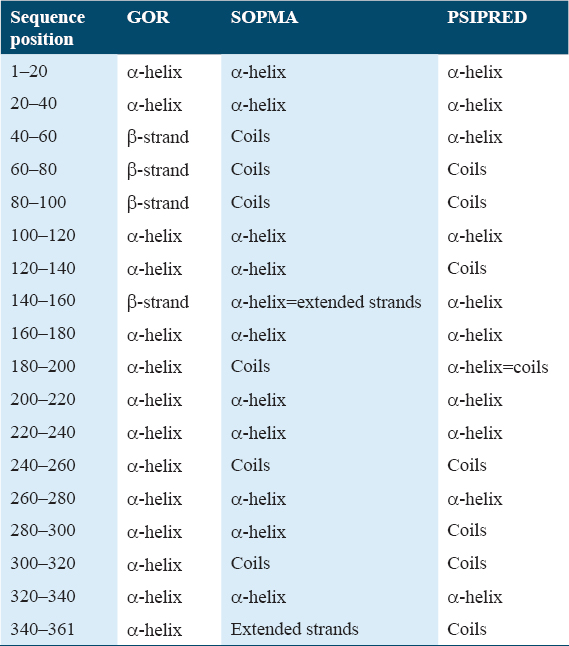
The Universal Protein Resource[6] database was used to extract the sequence of the protein RSAD2 (Accession number: Q8WXG1(RSAD2 Human)) and kept for the secondary structure analysis using Garnier-Osguthorpe-Robson (GOR),[7] Self-Optimized Prediction method with Alignment (SOPMA),[8] and Position-Specific Iterative-Blast based secondary structure prediction (PSIPRED).[9]
Modeling of protein structure
SWISS-MODEL[10] and Protein Homology/Analogy Recognition Engine (Phyre2)[11] servers were used to estimate the 3D-structure of the protein of interest, RSAD2. SWISS-MODEL is a server for automated three-dimensional protein structure comparison modeling. Its objective is to make protein modeling accessible and thus assisting all life science researchers of different domains across the world. The Phyre2 server is used to predict protein structure, function, and mutations. It’s a web-based service for predicting protein structure. Sequence template alignments are generated by them. Using the template structures, a good number of models were created, and the finest model was chosen by comparing their Z scores. A comprehensive search of the protein data bank (PDB) was conducted to pick out the potentially correlated sequences for which experimentally determined structures are previously known.
Validation of structures
Ramachandran plots were generated using the SAVES 6.0[12] software to validate predicted protein structures using parameters such as favored regions, allowed areas, and generously allowed regions of amino acid residues.[13] The PDB files of the best models of the target genes predicted by SWISS-MODEL and Phyre2 servers were uploaded to the SAVES 6.0 server to develop Ramachandran plots. They were made for predicted models, and the plots were compared to see which model was the most effective and stable.
Energy minimization of predicted structure
SPBDV (Swiss PDB viewer)[14] was used for energy minimization to achieve the lowest energy conformation for predicted and evaluated structures.[15]
MD simulation
The model with lowest energy was considered for MD simulations. iMODS was used to understand the best model stability in terms of the torsion angles between molecules.[16]
Results
Prediction of protein secondary structure
Tools such as GOR, SOPMA, and PSIPRED were used to estimate the secondary structure of RSAD2. They provided information on secondary structures such as α-helix, β-strand, and coils for target RSAD2. The GOR method is a secondary structure prediction methodology based on information theory. It is based on empirical studies of known protein tertiary constructions solved by X-ray crystallography. RSAD2 had 142 alpha helices, 74 extended strands, and 145 random coils, according to GOR analysis. SOPMA is a server that predicts protein structure, using consensus prediction from many alignments, the server led to significant advancements in protein secondary structure. RSAD2 had 146 alpha helices, 80 extended strands, 19 beta turns, and 116 random coils, according to SOPMA analysis.[17] PSIPRED is a protein structure forecasting system that allows users to enter a protein sequence, perform a prediction and receive the results in graphic form. PSIPRED analysis revealed that RSAD2 had 154 a- helices and 162 coils and 145 extended strands.
Template identification
identificationFor RSAD2 comparative homology modeling, a PDB blast was performed to find out the template structures.[17] The templates were compared, and the following are the final parameters [Table 2].

Protein structure modeling with SWISS-MODEL
MODELThe structures of the protein RSAD2 were modeled using the SWISS-MODEL program. For the RSAD2, 30 templates were produced using the SWISS-MODEL. The best two models are 6q2p [Figure 2a] and 5vsl [Figure 2b] based on alignment coverage and Z score.
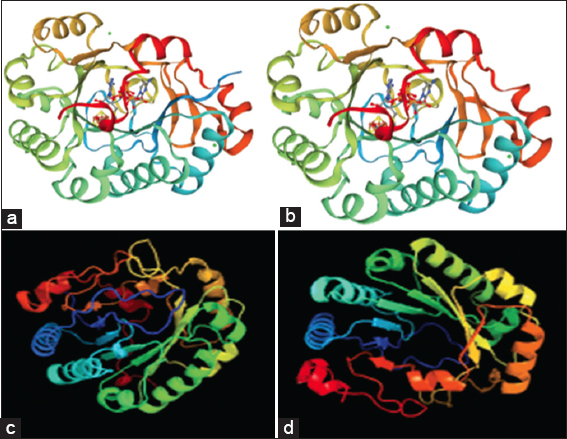
- Best predicted models for SWISS-MODEL (a and b) and Phyre2 (c and d)
Protein structure modeling using Phyre2 server
serverThe three-dimensional models and ligand binding sites were predicted using the Phyre2 server. Through Phyre2 server we got 114 templates in which 20 are with models and remaining are not modeled. Based on the maximum coverage, the best templates with models obtained are 6q2p [Figure 2c] and 5vsl [Figure 2d].
Structure validation
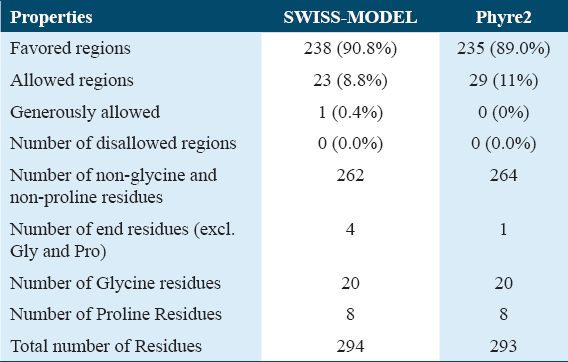
validationThe best predicted model of RSAD2 by SWISS-MODEL and Phyre2 was submitted to the online database SAVES 6.0, which generated the Ramachandran plot, to investigate the predicted models.[18] The amino acids (residues) were distributed in three zones. The three unique regions were favored regions, allowed regions, and generously allowed regions [Figure 3 and Table 3].
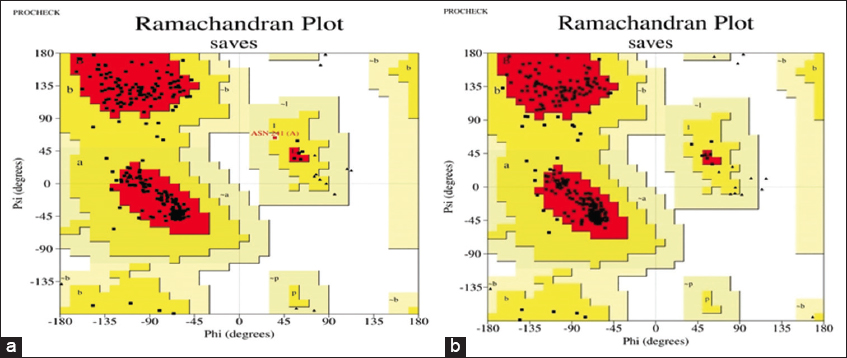
- Ramachandran plot analysis for SWISS-MODEL (a) and Phyre2 (b) predicted protein structures
Energy minimization
SPBDV[9] software carried out the energy minimization of the projected models. To determine the best projected models, the energy minimization values were compared. The least energetic model was represented by SWISS-MODEL (E=-18439.475KJ/mol) [Table 4].

MD simulation
After comparing the energy minimization outcomes, we can see that the best predicted protein model (Swiss-Model) has the lowest value for minimization.[19] As a result, the SWISS-MODEL was further interpreted for MD simulation to examine the stability and physical activities utilizing the iMODS webserver’s normal mode analysis (NMA).[20] Even with significantly large macromolecules, iMODS enabled the exploration of such modes and produced plausible transition paths between two homologous structures.[21] The unique internal coordinate formulation increases NMA’s efficiency and broadens its applicability while maintaining stereochemistry implicitly.[22]
The motion stiffness is expressed by the eigenvalue linked with each normal mode. The energy that is required to distort the structure determines its value.[13] The easier the deformation, the lower the eigenvalue. The calculated Eigen value for the predicted 3-D structure was 8.454e-04 [Figure 4a]. The eigenvalue is inversely proportional to the variation associated with each normal mode. Colored bars show the individual (red) and cumulative (green) variances [Figure 4b]. The covariance matrix shows how correlated (red), uncorrelated (white), or anti-correlated (blue) movements are experienced by pairs of residues [Figure 4c]. An elastic network model was built to depict the set of atoms connected by springs.[23] Each dot in the graph symbolizes a spring between the associated pair of atoms, with stiffer springs symbolized by darker grey dots and vice versa.[24] All above-mentioned points denote the higher stability of the complex [Figure 4d].
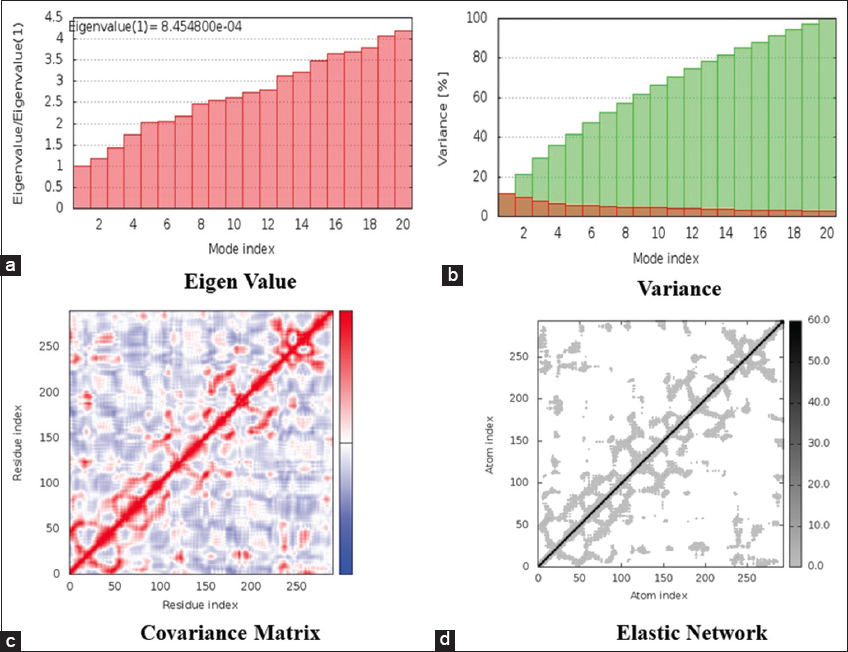
-
In silico simulation of SWISS-MODEL predicted 3-D structure of RASD2. (a) Eigenvalue (b) Variance (c) Covariance matrix (d) Elastic network
Discussion
In humans, the RSAD2 gene codes for a protein called radical s-adenosylmethionine. It is an interferon-inducible antiviral protein that is important in the antiviral state of the cell. Radical-SAM enzymes catalyze a variety of processes in nearly every area of cellular functioning. RSAD2 is an excellent illustration of an uncharacterized radical-SAM enzyme, and chemists and biologists have been trying to figure out what its substrates and biochemical processes are for nearly two decades. Studies on RSAD2’s antiviral action have led to a multifunctionality perspective since its discovery. RSAD2 inhibits virus reproduction by a variety of ways, including its radical-SAM catalytic activity or interactions with various viral or host proteins.[25-27] Since the 3D structure for the complete human RSAD2 is not available in the PDB, the complete RSAD2 protein structure needs to be modeled using different tools based on homology modeling. Homology modeling is one of the fundamental discoveries that precipitated a quick paradigm shift in computational biology. It obtains the three-dimensional structure of a target protein based on the similarity between template and target sequences. This method is useful for studying viral proteins that are difficult to crystallize, also it provides a deeper understanding of receptor-ligand interaction.[28] The structure of RSAD2 was predicted using the SWISS-MODEL and Phyre2 servers. The validation plays an important role in any study; therefore, the foremost model was validated by implying Ramachandran Plot which represented 98.8 % of stable amino acids in helices and Beta sheets as allowed regions. Finally, the optimal model was determined using energy minimization. One can interpret the stability of model based on the energy minimization, lower the energy; more stable the model reference, thus, model with the lowest energy was used further which was from SWISS-MODEL. It had an energy of −18439.475KJ/mol. Further MD simulations were done for the results of SWISS-MODEL and it was noted that it shows higher stability of the complex. Further studies can be progressed for looking an active site in the target protein and molecular modeling research to identify the appropriate substrate for RSAD2.
Conclusion
Using various in silico methods and approaches, the present preliminary inquiry aimed to comprehend the primary, secondary, and tertiary structures of the RSAD2 structural protein. The SWISS-MODEL outperformed the two anticipated models (Phyre2 and SWISS-MODEL) in Ramachandran plot validation. 238 residues (90.8%) were detected in the favored zone, 23 residues (8.8%) in the allowed region, and 1 residue (0.4%) in the generously allowed region during Ramachandran plot validation. The two models when subjected to energy minimization calculations using SPBDV, the SWISS-MODEL represented the lowest energy (E = −18439.75KJ/mol). Further the minimized structure was also subjected with MD simulations which represented a stable conformation. This study provided molecular insight for future investigations to identify the structural and functional features of RSAD2 protein to discover or build novel antiviral medicines against Zika virus using a structure-based drug design strategy.
Recommendations
Based on homology structure confirmation, validation, and dynamics, the RSAD2 three-dimensional structure was built. The suggested model could be relevant in researching RSAD2’s pathophysiological role in Zika virus infection.
Ethics Approval and Consent to Participate
None.
Availability of Data and Material
The data that support the findings of this study are openly available.
Competing Interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- A review on Zika virus outbreak, epidemiology, transmission and infection dynamics. J Biol Res (Thessalon). 2020;27:5.
- [Google Scholar]
- ddhCTP produced by the radical-SAM activity of RSAD2 (viperin) inhibits the NAD+-dependent activity of enzymes to modulate metabolism. FEBS Lett. 2020;594:1631-44.
- [Google Scholar]
- Prioritization of potential vaccine candidates and designing a multiepitope-based subunit vaccine against multidrug-resistant Salmonella Typhi str. CT18:A subtractive proteomics and immunoinformatics approach. Microb Pathog. 2021;159:105150.
- [Google Scholar]
- Nucleic Acids Res. 2015;43:D204-12.
- GOR V server for protein secondary structure prediction. Bioinformatics. 2005;21:2787-8.
- [Google Scholar]
- SOPMA:Significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Comput Appl Biosci. 1995;11:681-4.
- [Google Scholar]
- SWISS-MODEL:An automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381-5.
- [Google Scholar]
- The Phyre2 web portal for protein modeling, prediction and analysis. Nat Protoc. 2015;10:845-58.
- [Google Scholar]
- TopPred II:An improved software for membrane protein structure predictions. Comput Appl Biosci. 1994;10:685-6.
- [Google Scholar]
- Implying analytic measures for unravelling rheumatoid arthritis significant proteins through drug-target interaction. Interdiscip Sci. 2016;8:122-31.
- [Google Scholar]
- Antiviral phytocompounds target envelop protein to control Zika virus. Comput Biol Chem. 2018;77:402-12.
- [Google Scholar]
- GPU-BLAST:Using graphics processors to accelerate protein sequence alignment. Bioinformatics. 2011;27:182-8.
- [Google Scholar]
- Comparative modeling, characterization and energy minimization studies of aquaporin 9:An exclusive target protein for rheumatoid arthritis. Int J Pharm Invest. 2019;9:43-6.
- [Google Scholar]
- iMODS:Internal coordinates normal mode analysis server. Nucleic Acids Res. 2014;42:W271-6.
- [Google Scholar]
- Evasion of innate and intrinsic antiviral pathways by the Zika Virus. Viruses. 2019;11:970.
- [Google Scholar]
- Molecular modeling and network based approach in explaining the medicinal properties of Nyctanthes arbortristis, Lippia nodiflora for rheumatoid arthritis. J Bioinform Intell Control. 2014;3:31-8.
- [Google Scholar]
- Analysis of inhibitory activity of 2, 4-diamino-5-methyl-5-deazapteridine (DMDP) derivatives against recombinant human dihydrofolate reductase by molecular modeling. Online J Bioinform. 2011;12:357-70.
- [Google Scholar]
- Network based approach for understanding the chondrocyte biology for rheumatoid arthritis. Int J Innov Res Comput Commun Eng. 2014;2:332-7.
- [Google Scholar]
- Recent advances in hydrogels for biomedical applications. Asian J Pharm Clin Res. 2018;11:62-8.
- [Google Scholar]
- A unifying view of the broad-spectrum antiviral activity of RSAD2 (viperin) based on its radical-SAM chemistry. Metallomics. 2018;10:539-52.
- [Google Scholar]
- The first case of myxoinflammatory fibroblastic sarcoma affecting the maxillary bone. Int J Health Sci (Qassim). 2022;16:37-9.
- [Google Scholar]
- Examining the relationship between oral health promoting behaviour and dental visits. Int J Health Sci (Qassim). 2019;13:40-3.
- [Google Scholar]
- Next-generation sequencing technology in the diagnosis of mitochondrial disorders. Int J Health Sci (Qassim). 2020;15:1-2.
- [Google Scholar]




