
Translate this page into:
XXXYY variant of Klinefelter syndrome: A case report
Address for correspondence: Dr. Noorhan Moussa Rhayel, Road 2904, Salmaniya Medical Complex, Manama, Kingdom of Bahrain. E-mail: noorhan.46@live.com
This is an open-access article distributed under the terms of the Creative Commons Attribution-Noncommercial-Share Alike 3.0 Unported, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
This article was originally published by Qassim Uninversity and was migrated to Scientific Scholar after the change of Publisher.
Abstract
This case report is about a 19-month-old boy, product of an in vitro fertilization twin pregnancy and born to young non-consanguineous parents, who presented with speech and motor developmental delay. On genetic evaluation, he was found to have the exceedingly rare variant 49, XXXYY of Klinefelter syndrome. Given the rarity of this condition and the limited literature available, this case report will surely add value to the literature.
Keywords
Rare
chromosome aneuploidy
genetic disease

Introduction
Klinefelter Syndrome (KS) has a prevalence of approximately in 660 live born males and is known to have the karyotype of 47, XXY.[1] Other variants described in the literature include mosaic KS (46,XY/47,XXY), 48,XXYY, and 49,XXXYY. These additional sex chromosomes may possibly lead to the deleterious mental and physical outcomes found in patients.[2,3]
The variant 49,XXXYY was first reported at 1963 in a young male with intellectual disability and features similar to KS.[4] This variant is extremely rare, with a prevalence being estimated to be < 1 in one million. Patients typically have intellectual disability, autism, distinctive facial features, cryptorchidism, hypogonadism, skeletal malformations, and others.[5] The proposed mechanism is a nondisjunction that takes place during the formation of gametes or at conception.[6]
A few cases of the variant 49, XXXYY have been reported in the literature.[6] The literature was limited to case reports that were mostly published in the previous century.[3,4,7-10] Hence, the case presented in this report is considered to be a significant addition to the literature, as it is the most recent case report highlighting this rare variant.
Case Presentation
The 19-month-old patient presented with developmental delay, including decreased sucking reflex, inability to stand or walk without support, and speech delay, in comparison to his twin. Antenatal history was uneventful and prenatal DNA study showed a normal karyotype, which is done routinely in those who conceive through in vitro fertilization. As a product of in vitro fertilization twin pregnancy, he was delivered at 36 weeks with a birthweight of 1.5 kg by elective lower segment cesarean section. The patient was admitted to the neonatal intensive care unit for 10 days due to his low birth weight and decreased sucking reflex, completed uneventfully. Bilateral cryptorchidism was noted and operated twice (one for each testis) at 3 and 18 months of age. The birth weight of the twin was 2.5 kg with an uneventful postnatal history. His weight is 12 kg (at 50.798 percentile) with a height of 77 cm (at 2.442 percentile) and his milestones are appropriate for his age. In vitro fertilization was done due to the paternal oligozoospermia, with the mother being free from any medical illnesses. The parents lacked dysmorphic features and were young upon conception; the father was 28 years old and the mother was 25 years old. In addition, no family history of any congenital anomalies was present in either parent.
On physical examination, the patient was 73 cm (at 0.1 percentile) tall and weighed 11.3 kg (at 29.46 percentile). He had prominent forehead and mild hypertelorism with wide nasal bridge [Figure 1], micrognathia with macrodontia [Figure 2], and bilateral ear creases [Figure 3]. Genital exam revealed small testis with micropenis. Other features were notably musculoskeletal, which were postural lumbar kyphosis (There was difficulty obtaining picture of Kyphosis), bilateral forearms fixed in mid-prone position with limitations in supination and pronation but with a full flexion and extension of the elbow, left fifth digit clinodactyly [Figure 4], right varus knee deformity, bilateral pes planus [Figure 5], and an out toeing gait. Wrist and hip examination were normal, as well as other systems.
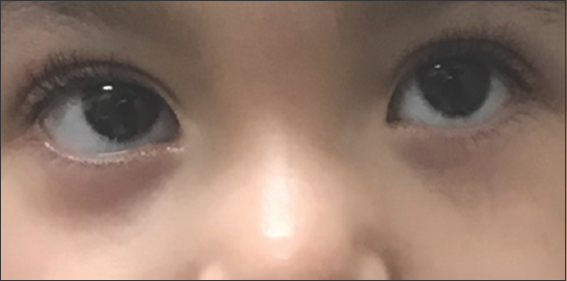
- Hypertelorism with wide nasal bridge
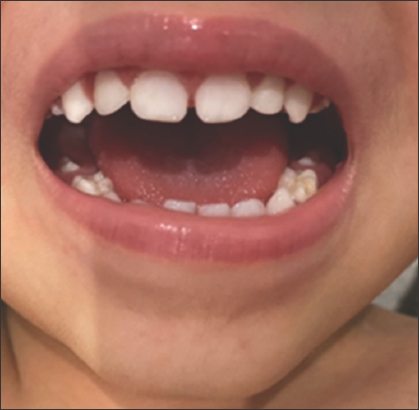
- Micrognathia with macrodontia. The image was taken recently, at the age of approximately 2 years and 10 months
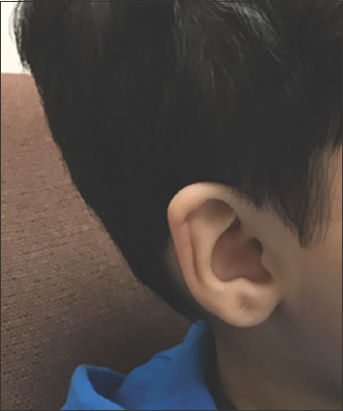
- Ear crease

- Left 5th digital clinodactyly
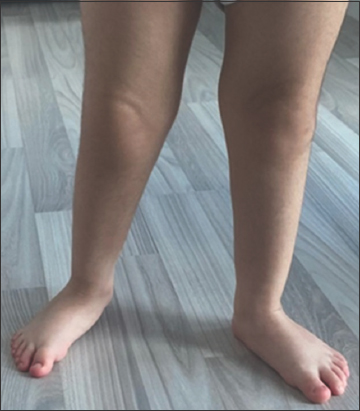
- Right varus knee deformity and bilateral pes planus
Laboratory investigations
Laboratory investigations done to rule out other causes of delayed milestones were all normal, including hemoglobin, iron profile, Vitamin D, renal, and liver function tests.
Cytogenetics studies
A standard karyotype was performed, revealed an abnormal male karyotype of two additional copies of X and one additional copy of Y chromosome in all analyzed metaphases [Figure 6]. Thus, 49, XXXYY variant of KS was diagnosed. Furthermore, parental standard karyotype was found to be normal, which ruled out numerical and/or structural chromosomal anomalies in the parents such as paternal KS [Figures 7 and 8].
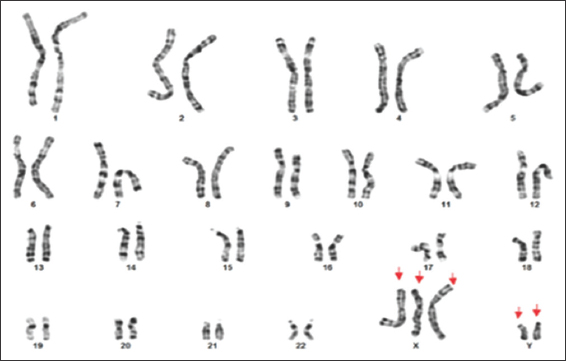
- Patient’s Karyotype, XXXYY pattern
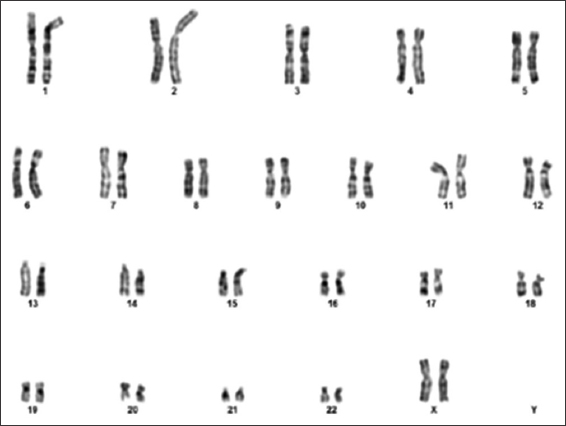
- Maternal karyotype showing normal 46, XX
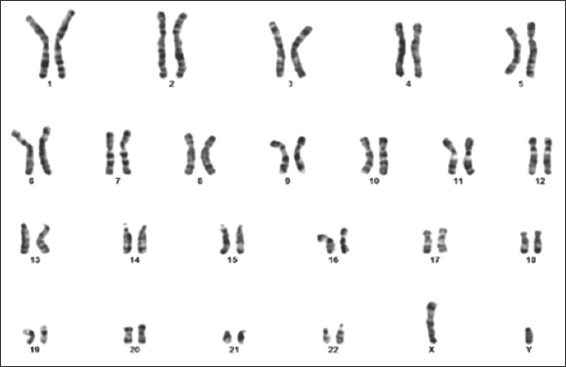
- Paternal karyotype showing normal 46, XY
Radiological studies
Forearm X-ray revealed bilateral proximal cartilaginous radioulnar synostosis [Figure 9], which then progressed to a bony type on the newer X-Ray [Figure 10]. X-ray of the spine and hips showed spina bifida in addition to bilateral mild hip acetabular dysplasia with acetabular index of 24.2° at the left and 23° at the right [Figure 11].
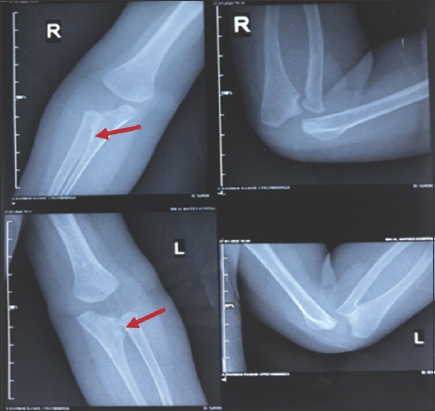
- Previous X-ray of both forearms shows bilateral proximal radioulnar cartilaginous synostosis, as highlighted by the red arrows. This X-ray was done when the patient was approximately 7 months of age
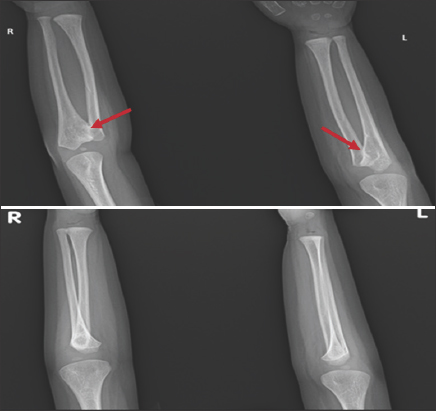
- Newer X-rays of both forearms shows bilateral proximal radioulnar synostosis of bony type, as highlighted by the red arrows. This X-ray was done when the patient was approximately 2 years and 10 months old
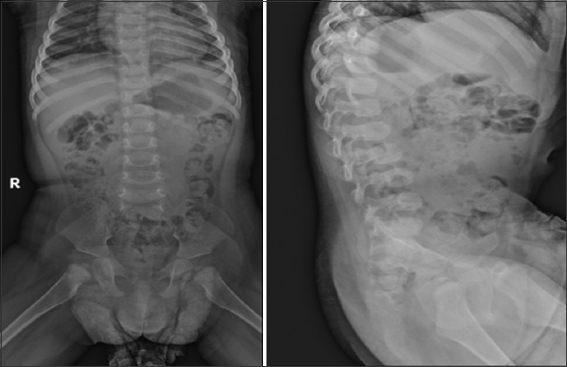
- Hip and spine X-rays. Hip X-Ray shows bilateral mild hip acetabular dysplasia with acetabular index of 24.2° at the left and 23° at the right. Spine X-ray shows spina bifida. This X-ray was taken when the patient was approximately 20 months old
Discussion
The manifestations of our patient with the other confirmed XXXYY patients are summarized in Table 1. Three case reports presented patients who are relatively close to our patient’s age.[3,4,8] Facial features, genital abnormalities, delayed milestones, and intellectual disability were similar with minor variations between the cases. Our patient was noticed to have certain facial and musculoskeletal features such as micrognathia, wide nasal bridge, bilateral hip dysplasia, and an out toeing gait which may have gone unnoticed or were absent in other patients reported by the case reports.[1-11] Clinodactyly was reported by us and by Benn et al. only.[8] Our patient had radioulnar synostosis which was only seen in a mosaic 48,XXYY/49,XXXYY patient reported by Salamanca et al.[9] It is crucial to note that case reports of adult patients do show some features that are age related, such as scantiness or absence of facial hair, which may be seen in our patient when he advances in age.[7,9,10]
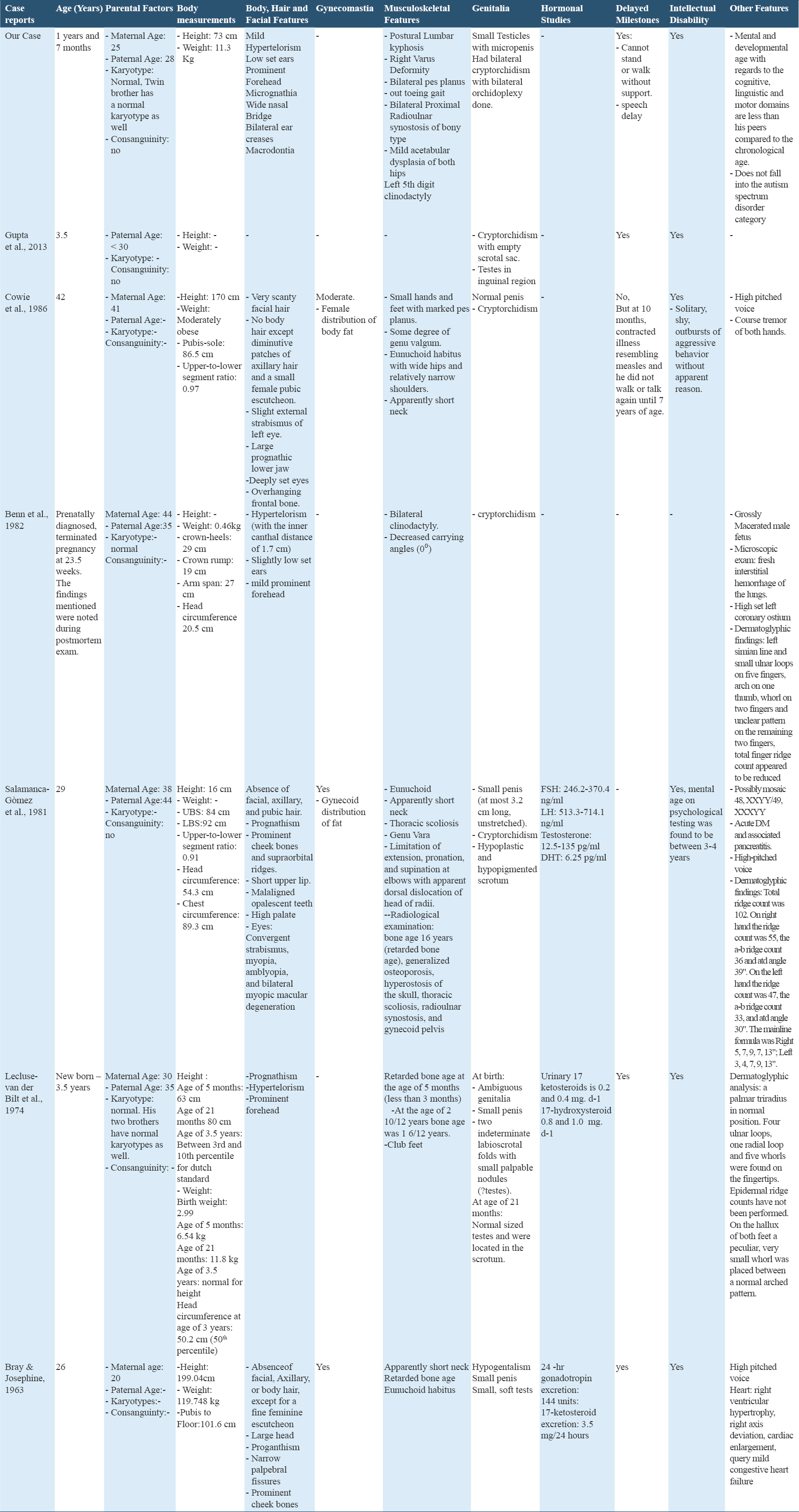
The extra sex chromosomes attribute to the mental and physical development of such cases. The proposed mechanisms by which this aneuploidy occurs are either: Fertilization of a normal ovum with one X by an XXYY sperm which arose from two paternal meiotic non-disjunctions at both meiotic divisions, fertilization of an XX ovum by an XYY sperm, or fertilization of an XXX ovum by a YY sperm.[8] For each possibility for the origin of the polysomy, a minimum of two independent non-disjunctions must be postulated; at least one of which is paternal in origin. In our case, parents and sibling had normal karyotypes, similar to what Lecluse-van der Bilt et al. reported.[4] Gupta et al. reported a patient who had a young, non-consanguineous parents, similar to our patient.[3] This may suggest that other unknown factors exist.
49,XXXYY variant is extremely rare with only two cases reported in the 21st century, with our patient being the third. Our case report presents a unique finding of XXXYY syndrome in a patient who was an IVF product and a twin of a normal sibling who had a normal prenatal DNA karyotype. The future studies may shed light upon additional features as well as the possible outcomes and complications of this variant.
Author’s Declaration Statement
Ethical approval
Not applicable.
Declaration of patient consent
Written informed consent has been obtained from the father of the studied patient and not the patient himself.
Data availability statement
All data are available in the manuscript.
Competing interest
The author reports no conflicts of interest.
Funding Statement
None.
Authors’ contributions
-
Dr. Ali Alekri Case recognition
-
Dr. Maryam Busehail Supervisor and editor
-
Dr. Noorhan Moussa Rhayel Data collection, data interpretation, discussion and conclusion
-
Dr. Sayed Mohamed Almosawi Data collection, data interpretation, discussion and
Acknowledgments
The authors acknowledge the patient’s parents for their great co-operation.
References
- Klinefelter syndrome mosaicism 46,XX/47,XXY:A new case and literature review. J Pediatr Genet. 2020;9:221-6.
- [Google Scholar]
- Multiple XY syndrome:A case study. Int J Res Appl Nat Soci Sci (IMPACT:IJRANSS). 2013;1:87-90.
- [Google Scholar]
- 49, XXXYY Syndrome Orphanet. Available from: https://www.orpha.net/consor/cgi-bin/oc_exp.php?lng=en&expert=261534
- Genetic and Rare Diseases Information Center (GARD)-an NCATS Program. Rarediseases.info.nih.gov. https://www.rarediseases.info.nih.gov/diseases/10922/49/xxxyy/syndrome#:~:text=49%2C%20XXXYY%20syndrome%20is%20a,reported%20in%20the%20medical%20literature
- 49, XXXYY chromosome anomaly in a mentally retarded man. Br J Psychiatry. 1986;148:210-2.
- [Google Scholar]
- An XXXYY sex-chromosome anomaly. Report of a mentally deficient male. JAMA. 1963;184:179-82.
- [Google Scholar]
- First report of two rare entities in a family:49,XXXXY and 45,X. J Pediatr Genet. 2017;6:174-6.
- [Google Scholar]




